
Л. В. Щербакова практикум по аналитической химии барнаул 2004 министерство образования российской федерации алтайский государственный университет химический факультет практикум
| Вид материала | Практикум |
- Практикум Владивосток 2004 Министерство образования и науки Российской Федерации Владивостокский, 628.95kb.
- Практикум Ярославль, 2004 Министерство образования и науки РФ ярославский государственный, 885.94kb.
- Министерство Образования Российской Федерации Московский Государственный Университете, 1997.23kb.
- А. М. Горького Кафедра алгебры и дискретной математики Щербакова В. А. Лабораторный, 418.72kb.
- Анализ воды, 505.46kb.
- Российской Федерации Министерство образования и науки Российской Федерации Государственный, 343.55kb.
- Учебно-практическое пособие Хабаровск, 2001. Министерство образования Российской Федерации, 2795.36kb.
- Практикум ростов-на-Дону 2008 министерство образования и науки российской федерации, 1352.33kb.
- Учебное пособие Министерство общего и профессионального образования Российской Федерации, 936.13kb.
- Российской Федерации Уральский Государственный Университет им. А. М. Горького Философский, 1670.87kb.
II. Иодометрическое определение меди
Реагенты:
Стандартизованный раствор тиосульфата натрия;
Иодид калия, 20%-ный раствор;
Серная кислота, 4 М раствор;
Индикатор – крахмал.
Выполнение определения
В мерную колбу вместимостью 100,0 мл получают у преподавателя раствор, содержащий соль меди, разбавляют дистиллированной водой до метки и хорошо перемешивают. Бюретку заполняют стандартизованным раствором тиосульфата натрия. В колбу для титрования мерным цилиндром отмеривают 5 – 7 мл раствора KI (20%), 8 – 10 мл раствора серной кислоты, аликвоту (10,00 мл) анализируемого раствора меди и оставляют смесь на 5 мин в темном месте. Затем приступают к титрованию. Крахмал прибавляют, когда окраска раствора станет светло-желтой. Синяя окраска должна исчезнуть от одной капли раствора тиосульфата натрия и не появляться в течение 2 – 3 минут. В результате реакции образуется малорастворимый иодид меди (I). Осадок иодида меди должен иметь цвет слоновой кости. Проводят не менее 3-х параллельных определений. Рассчитывают содержание меди и проводят статистическую обработку результатов.
Расчет результатов анализа:
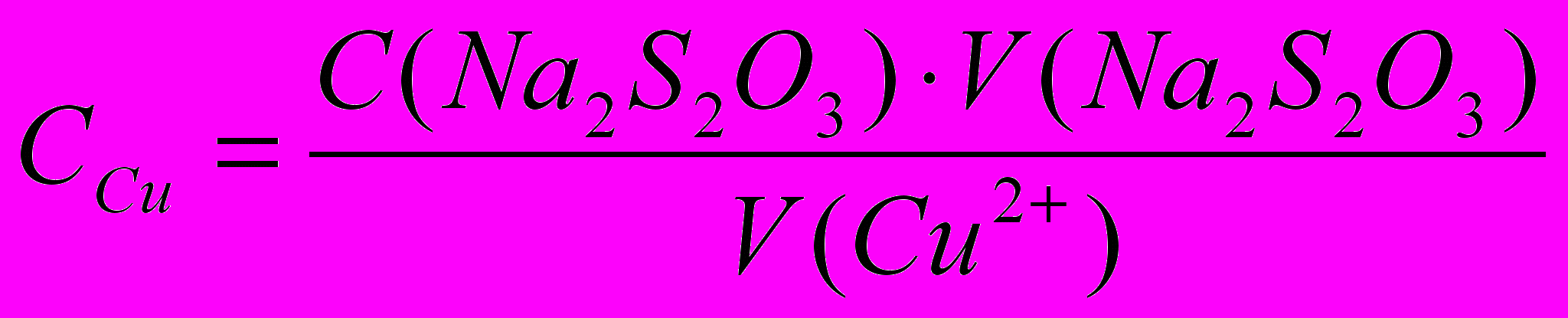
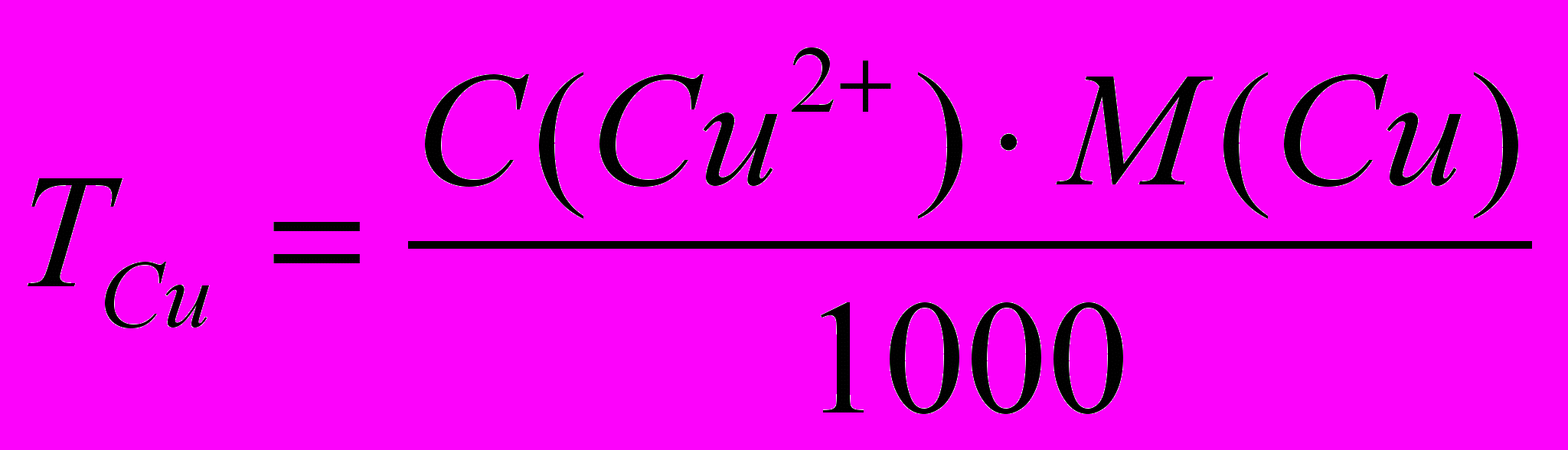
mCu = Tcu Vm.k.
Г Р А В И М Е Т Р И Ч Е С К И Й А Н А Л И З
Основные теоретические положения гравиметрического анализа (гравиметрии)
Сущность гравиметрического анализа
Сущность гравиметрического анализа (гравиметрии) заключается в определении массы продукта химической реакции. Измеряют взвешиванием на аналитических весах массу вещества называемого гравиметрической формой.
Гравиметрия является наиболее простым, точным, хотя и продолжительным методом анализа. Метод широко применяют для количественного определения неорганических и органических веществ.
Гравиметрический анализ выполняют с использованием методов отгонки или осаждения.
Метод отгонки
В этом методе определяемая часть анализируемого объекта должна быть летучей или должна превращаться в летучее соединение по той или иной химической реакции. Отгонка в свою очередь может выполняться как в прямом, так и в косвенном вариантах.
Примером прямого метода может служить определение СО2 в карбонатных породах. В этом случае навеску образца обрабатывают кислотой:
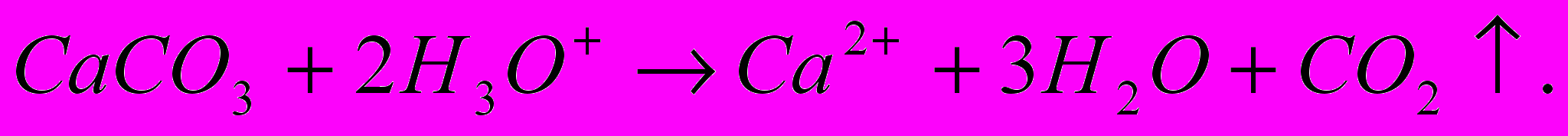
СО2 отгоняют в предварительно взвешенный сосуд, содержащий поглотитель (натронную известь – смесь NaOH, CaO, или аскарит – асбест, пропитанный NaOH). По увеличению массы сосуда судят о содержании СО2 в анализируемом образце.
В косвенном методе о содержании летучего компонента судят по убыли массы взятой навески анализируемого вещества. Таким способом часто определяют содержание воды. Для этого навеску анализируемого образца высушивают при определенной температуре и по убыли массы судят о содержании воды. Естественно, что в анализируемом веществе должны отсутствовать другие, летучие при данной температуре, компоненты.
Метод осаждения
Наибольшее значение в гравиметрии имеет метод осаждения. При этом определяемый компонент выделяют из раствора в виде малорастворимого соединения (или в виде элемента), которое после соответствующей обработки взвешивают. Например, для определения содержания бария в растворе его осаждают серной кислотой в виде сульфата:
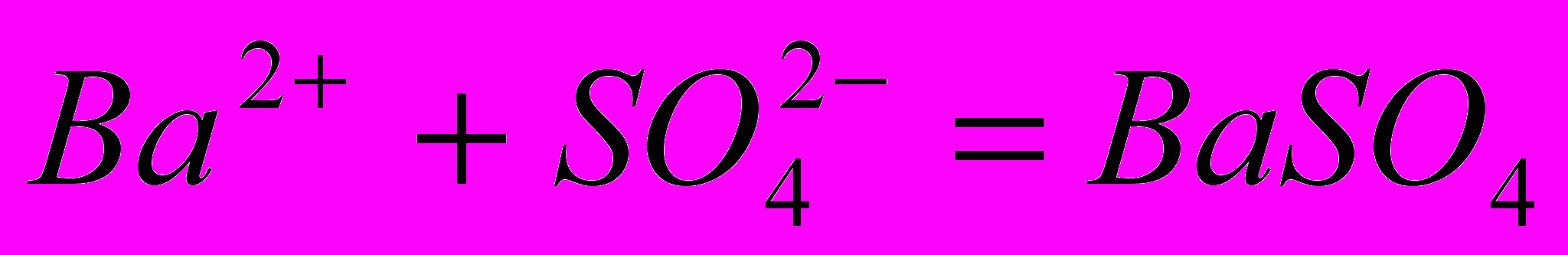 ↓
↓Осадок сульфата бария отделяют от раствора, промывают, прокаливают и взвешивают. По массе осадка рассчитывают содержание бария в растворе. Часто взвешивают не то соединение, которое осаждают, поэтому в гравиметрии различают осаждаемую и гравиметрическую формы, например:
| Определяемый элемент | Осаждаемая форма | Гравиметрическая форма |
| Cu | Cu | Cu |
| Ba | BaSO4 | BaSO4 |
| Al | Al2O3 nH2O | Al2O3 |
| Ca | CaC2O4 H2O | CaC2O4, CaCO3, CaO, CaSO4 |
Если химический состав осаждаемой формы может быть неопределенным, то основное требование к гравиметрической форме заключается в том, что она должна иметь постоянный состав, отвечающий определенной химической формуле. Таким образом, в гравиметрии имеют дело с малорастворимыми веществами. Не всякие осадки могут быть использованы в гравиметрии, к ним предъявляется ряд требований.
Требования к осадкам в гравиметрии
- Осадок должен быть практически нерастворимым, т.е. определяемый элемент (ион) должен выделятся в осадок количественно, (осаждение считается количественным, когда остаточная масса осаждаемого вещества остается за пределами точности взвешивания аналитических весов (0,0002 г.)).
- Осадок должен выделяться в форме, удобной для его отделения от раствора и промывания, он должен быть по возможности крупнокристаллическим или хорошо скоагулированным, если он аморфен. Важно, чтобы он был однородным по дисперсности (размерам частиц).
- Осадок должен быть чистым, по возможности не содержать посторонних примесей (аналитик должен иметь возможность оценивать его чистоту).
Растворимость осадков
Степень полноты осаждения определяется растворимостью осаждаемого вещества во время образования, фильтрования и промывания осадка. Растворимость малорастворимого соединения зависит от многих факторов. Основные из них: присутствие в осаждаемом растворе электролитов, кислотность раствора, присутствие комплексообразующих веществ, температура, природа растворителя.
Лабораторная работа№9
Определение содержания кристаллизационной воды в хлориде бария
Цель работы: освоить правила и технику работы на аналитических весах, изучить и освоить косвенный метод отгонки.
Сущность метода: определение разности массы кристаллогидрата и высушенной соли.
Аппаратура: аналитические весы, кл.2; сушильный шкаф.
Реактивы и посуда:
кристаллический хлорид бария;
бюкс;
эксикатор.
Выполнение определения:
Чистый сухой бюкс с крышкой взвешивают на технохимических весах с точностью до 0,05 г, затем с точностью ± 0,1 мг – на аналитических весах. Для этого на шлифованной поверхности вымытого и высушенного бюкса и крышки простым карандашом ставят номер. Затем необходимо внимательно изучить и в последующем строго выполнять правила работы на аналитических весах (например, брать бюкс и его крышку лишь пинцетом). После взвешивания и записи показаний весов, бюкс с крышкой, положенной боком на бюкс, помещают в сушильный шкаф и сушат при температуре 1200С 15 – 20 минут.
Горячий бюкс из сушильного шкафа вынимают пинцетом или тигельными щипцами (будьте осторожны, бюкс легко сломать!) и помещают в эксикатор на 10 – 15 минут для охлаждения. Крышку эксикатора придерживают большими пальцами, иначе она может легко соскользнуть и разбиться.
После охлаждения бюкс взвешивают на аналитических весах, так как его масса примерно уже известна. Взвешивание проводиться с точностью до ± 0,1 мг, данные записываются в рабочий журнал. Если разница между вторым и первым взвешиванием больше, чем 0,2 – 0,3 мг, то снова повторяют сушку, охлаждение в эксикаторе и взвешивание. И так до тех пор, пока разница между последними взвешиваниями будет не более 0,2 – 0,3 мг. Это означает, что масса бюкса с крышкой доведена до «постоянной массы» (масса “т”-тара)
Кристаллы хлорида бария измельчают в ступке до зёрен размером около 1 мм и с лопаточки (совочка) помещают в бюкс в количестве около 1г. Бюкс закрывают крышкой и взвешивают сначала на технохимических, а затем на аналитических весах с точностью ±1 мг (масса”б”- брутто), данные записывают в журнал.
При переносе бюкса в весовую комнату его нельзя брать руками и нагревать теплом рук. После взвешивания бюкс открывают, крышку кладут на него боком, ставят в сушильный шкаф. Сушат при температуре 1200С в течение 1 часа.
Горячий бюкс осторожно вынимают из шкафа и, не закрывая крышкой, охлаждают в эксикаторе 10 – 15 минут (крышку эксикатора закрывают через 5 – 7 секунд).
После охлаждения бюкс с солью в эксикаторе закрывают крышкой и взвешивают сначала на технохимических, а затем на аналитических весах, данные записывают в журнал. Открыв крышку, снова ставят бюкс с солью в сушильный шкаф на 20 – 30 минут, охлаждают в эксикаторе и взвешивают. Если разница в массе более 0,2 – 0,3 мг, то повторяют сушку, охлаждение и взвешивание до «постоянной массы»
(масса “m”).
Расчёт массового содержания воды (%) Xэкс. ведут, используя 5 значащих цифр, по формуле:
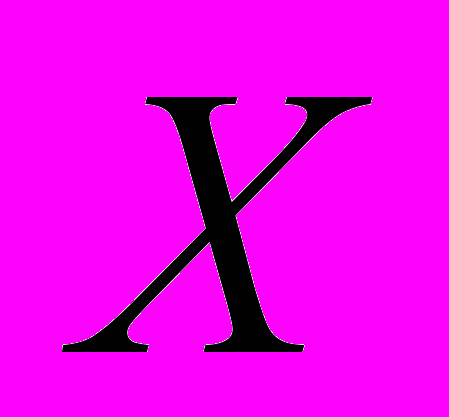 экс=
экс=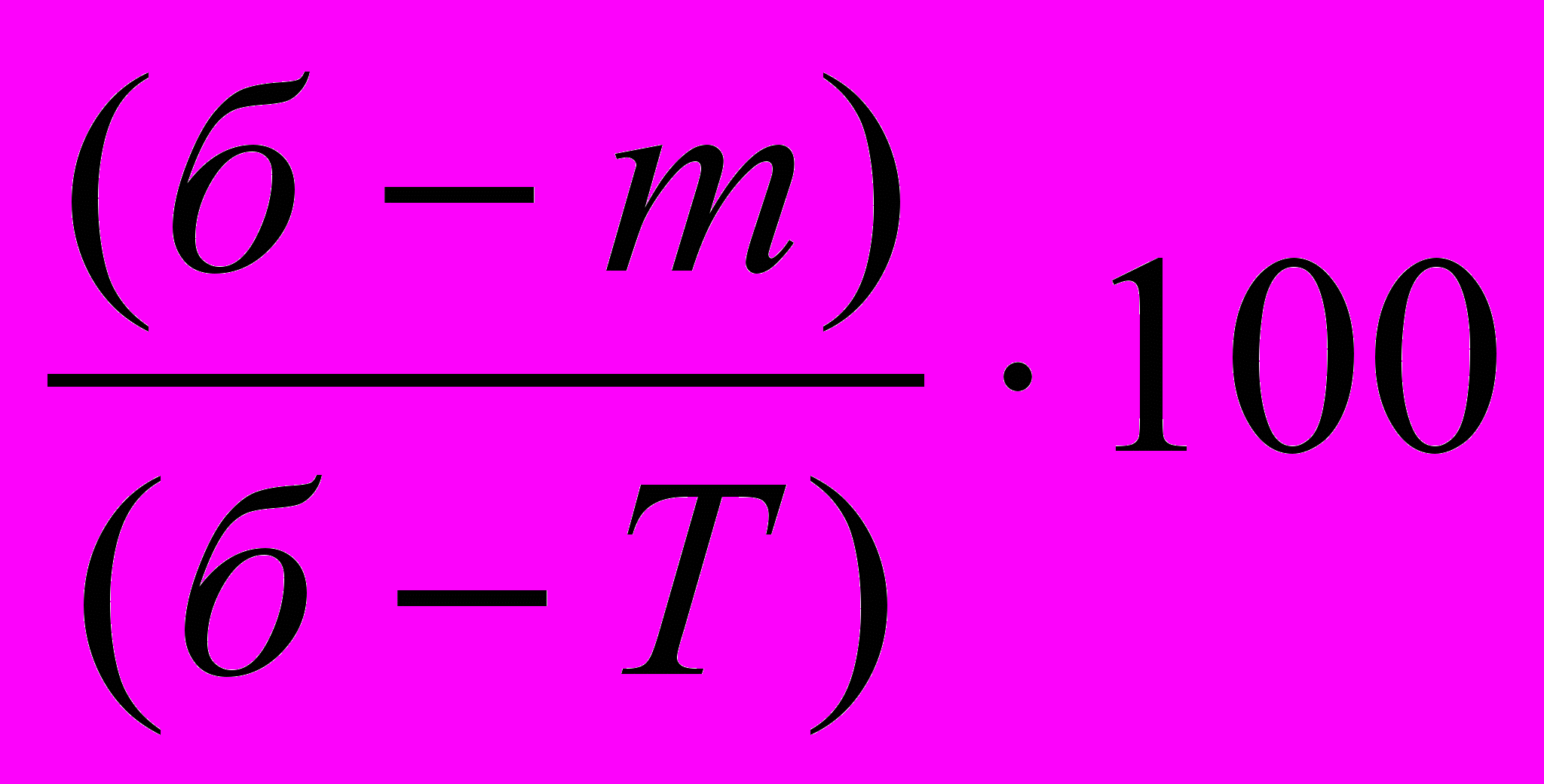 % =
% = 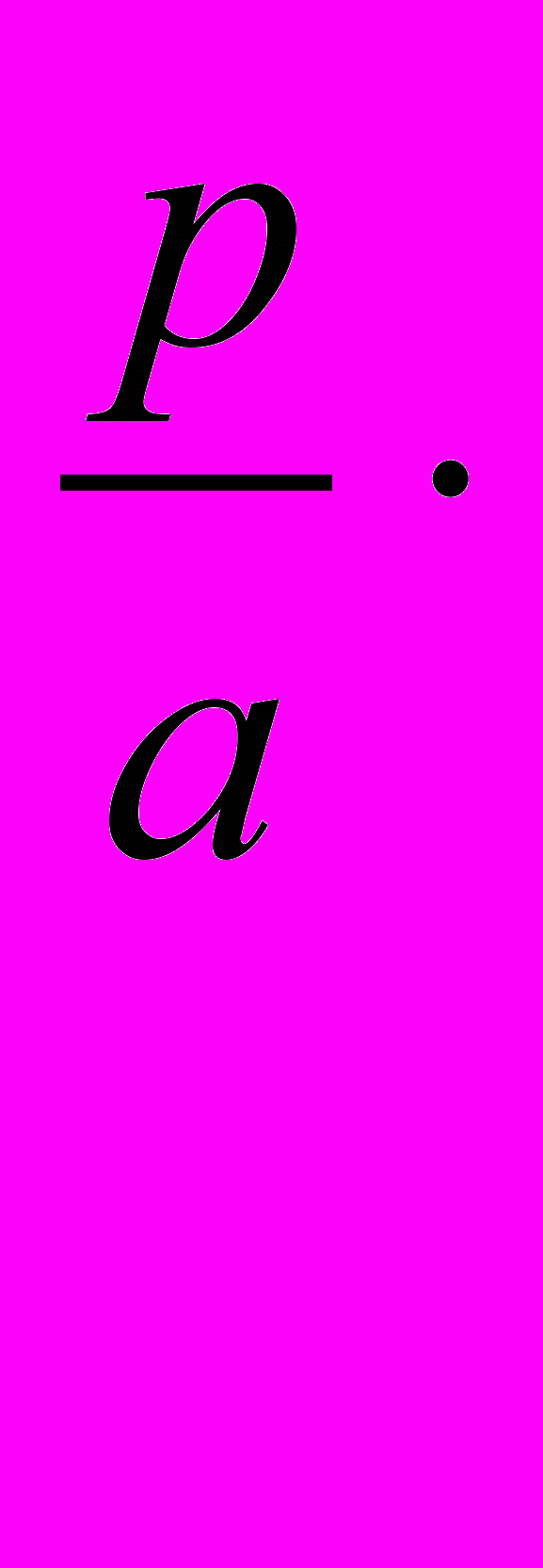 100%.
100%.где а=(б-Т) – масса кристаллогидрата, г,
р=(б-m) – масса воды, г.
Далее рассчитывают абсолютную ошибку между найденным массовым содержанием (%) воды в BaСl2*nH2O и теоретически вычисленным значением(BaСl2*2H2O):
∆X =Xэкс- Xтеор.
Затем рассчитывают относительную ошибку: ∆X. 100/a.
Отчёт. Кратко описывают: 1) сущность метода; 2)ход работы; 3)все результаты взвешиваний, даже если они совпадают; 4) расчёты процентного содержания воды и ошибок определения; 5) заключение (вывод)
Техника безопасности. При выполнении соблюдать следующие меры безопасности:
перед включением сушильного шкафа проверить его заземление;
2) горячие бюксы из сушильного шкафа брать пинцетом или тигельными щипцами;
3) после работы с солями бария тщательно вымыть руки.
Лабораторная работа №10
Определение содержания сульфат-ионов(серы)
Цель работы: освоить и приобрести навыки количественного анализа вещества по методу осаждения кристаллического осадка.
Сущность метода: образование малорастворимого кристаллического осадка ВаSO4 (ПР = 1,1 . 10-10) по реакции:
Ва+2 + SO4-2 = ВаSO4 ↓
Аппаратура, посуда, реактивы:
муфельная печь; аналитические весы, 2 кл.; фарфоровый тигель; эксикатор; стакан вместимостью 150 - 250 мл; воронка; бюретка; хлорид бария, ВаСI2, 0,2 М раствор; хлороводородная кислота, НСI, 2 М раствор; фильтр “синяя лента”:
Методика определения: Заранее прокаливают тигель и доводят его до «постоянной массы». Для этого взвешивают тигель сначала на технохимических, а затем на аналитических весах с точностью ± 0,1 мг и ставят его прокаливать в муфельную печь на 30 минут при температуре 800 0С. После этого тигель щипцами вынимают из печи, помещают в эксикатор на 20 – 30 мин для охлаждения (крышку эксикатора закрыть через 10 – 15 секунд), затем взвешивают с точностью ± 0,1 мг. Прокаливание, охлаждение, взвешивание повторяют до «постоянной массы» “т”.
У преподавателя получают контрольную задачу в стакане вместимость 150 мл. Предварительно стакан моют, ополаскивают дистиллированной водой (сушить стакан не надо), приклеивают этикетку с фамилией студента. В стакан добавляют 50 – 60 мл воды, 2 мл 2 М раствора НСI и нагревают на плитке до кипения (кипятить нельзя!).
Осаждение: Гравиметрическая форма совпадает с осаждаемой формой. Для получения более чистого крупнокристаллического осадка сульфата бария осаждение необходимо вести очень медленно, из разбавленного, кислого (рН<2), горячего раствора.
Промытую, чистую бюретку заполняют 0,2 М раствором ВаСI2. Из бюретки в стакан, в горячий раствор капают только одну каплю раствора ВаСI2, перемешивают раствор стеклянной палочкой не менее 5 с (нельзя касаться палочкой дна и стенок сосуда и вынимать ее). Затем капают вторую каплю раствора ВаСI2, непрерывно перемешивают раствор; через 5 с – 3 каплю и т.д. После 10 капель (около 0,5 мл) интервалы между каплями сокращают до трех секунд, после 20 капель – до двух, после 30 капель- до одной секунды. Затем их капают с такой скоростью, чтобы можно было сосчитать капли (примерно через 0,5 с). Необходимое количество осадителя: 15 – 20 мл. Время осаждения: 10 – 15 мин.
Стакан с осадком ставят на кипящую водяную баню на 10 – 15 минут (на плитку ставить нельзя, произойдет выброс осадка). После отстаивания осадка проверяют полноту осаждения: к прозрачному раствору приливают 2 – 3 капли раствора ВаСI2 (из бюретки). Если появится муть, то при перемешивании, по каплям добавляют еще 2 – 5 мл раствора ВаСI2, снова ставят на водяную баню на 10 – 15 мин. Если раствор прозрачный, не вынимая стеклянной палочки, закрывают стакан чистой бумагой и оставляют для созревания осадка до следующего занятия. На этом же занятии необходимо довести тигель до постоянной массы. Оставлять тигли в муфельной печи нельзя, их хранят в эксикаторе.
Рис. 1. Правильно сложенный бумажный фильтр
Рис. 2. Правильно вложенный в воронку фильтр
Фильтрование. Для фильтрования берут плотный фильтр («синяя лент») диаметром 7 - 11 см., сворачивают его, вставляют в чистую воронку (край воронки должен быть выше края фильтра на 1 см), смачивают дистиллированной водой из промывалки и плотно прижимают к стенкам воронки, особенно в местах, где находятся складки фильтра (рис.1, 2). Воронку с фильтром помещают в кольцо штатива и подставляют под нее чистый стакан, касаясь внутренней стенки стакана концом трубки воронки. Сначала декантируют прозрачную жидкость (рис.3), что ускоряет фильтрование в десятки раз.
Для этого осторожно снимают бумагу со стакана с осадком и вынимают палочку так, чтобы не взмутить осадок. Порцию прозрачной жидкости над осадком по палочке, медленно, чтобы не взмутить осадок, сливают из стакана на фильтр (декантация). Уровень жидкости в воронке должен быть на 0,5 см ниже края фильтра. Стакан с остатком раствора медленно приводят в более вертикальное положение, ставят на стол в наклонном положении (чтобы не взмутить осадок). Когда порция жидкости пройдет полностью через фильтр, снова по палочке, осторожно приливают на фильтр новую порцию прозрачной жидкости.
Промывание осадка. Когда прозрачная жидкость почти вся будет слита, к осадку в стакан прибавляют 5-7 мл дистиллированной воды. Осторожно взболтав осадок (не размазывать его по стенкам стакана), дают ему отстояться и сливают отстоявшуюся жидкость по палочке на фильтр. Такое промывание декантацией проводят 3-4 раза, давая возможность каждой порции жидкости полностью стекать. В последний раз взмученный осадок из стакана осторожно по палочке, по возможности сразу весь, переносят на фильтр. Остатки осадка в стакане полностью (в 2-4 приема) смывают тонкой струей воды из промывалки на фильтр. Здесь и затем при промывании осадка необходимо следить, чтобы содержимое воронки во всех случаях было на 0,5 см ниже края фильтра.
П
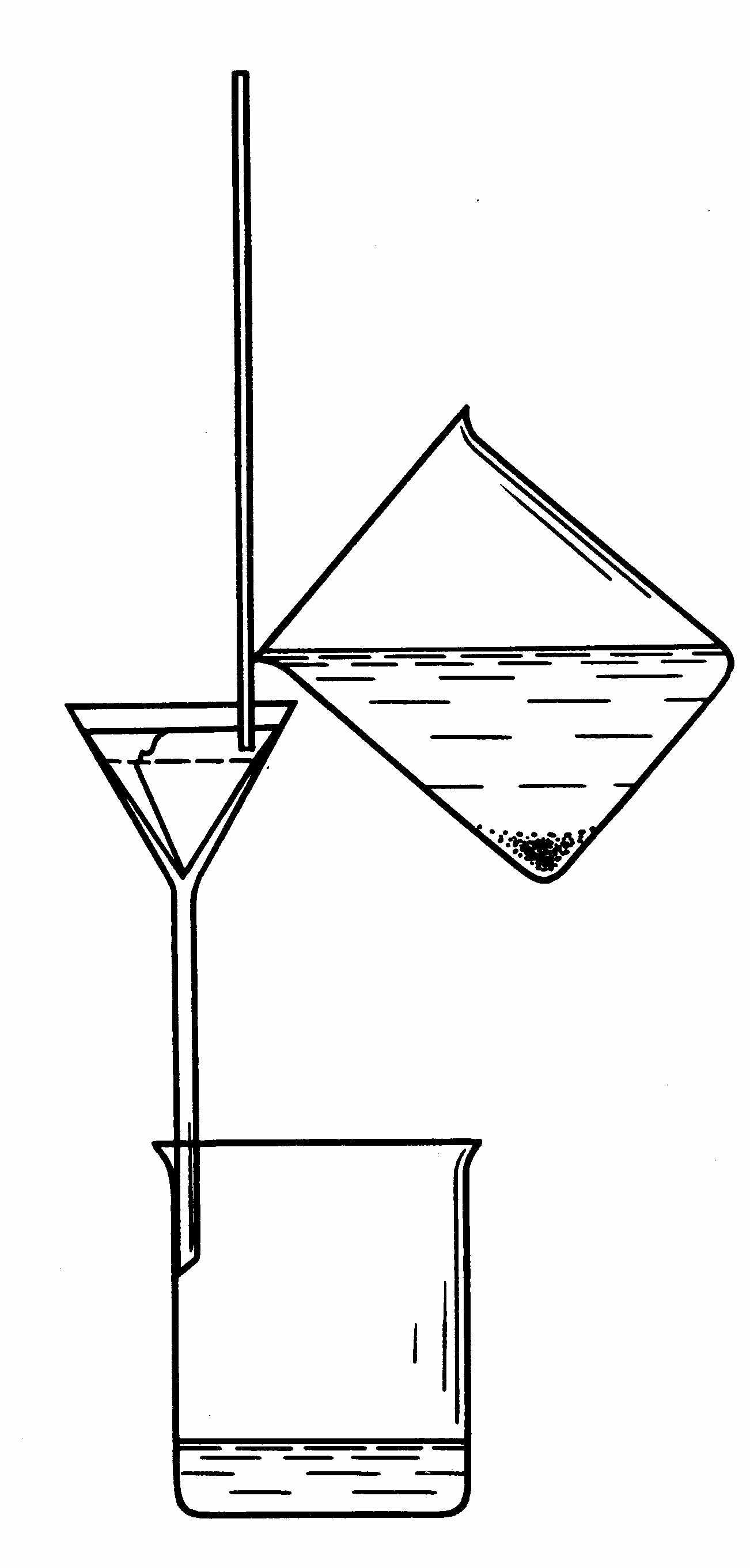
Рис. 3. Фильтрование через бумажный фильтр
ромывание осадка на фильтре проводят дистиллированной водой из промывалки, порциями по 20 мл. Для этого сначала тонкую струю воды осторожно направляют на свободную поверхность воронки, затем по кругу на верхнюю часть фильтра. Каждая порция жидкости должна полностью стекать. Промывка ведется 4-6 раз до отрицательной реакции на хлорид-ион: к порции фильтрата (около 0,5 мл), собранной в пробирку, добавляют 1 каплю 2 моль/л AgNO3. Мути не должно быть, возможна легкая опалесценция.
Объем фильтрата необходимо записать в журнале для последующего учета потерь осадка за счет его растворения. Обычно он составляет около 200мл =0.2 л.
Высушивание и прокаливание. Фильтр с осадком извлекают из воронки за край, где сложено 3 слоя, и слегка отжимают. Затем загибают края и помещают в тигель, предварительно доведённый до «постоянной массы» “т”. Тигель с фильтром ставят на 5-10 минут в сушильный шкаф при температуре 1200С, а затем с ещё влажным фильтром переносят в муфельную печь и ставят у её входа, приоткрыв дверцу. После того, как фильтр обуглится и истлеет (появление пламени не допускать!), тигель переставляют в самую горячую часть печи (осторожно! не уроните другие тигли). Прокаливание ведут 20 – 30 минут при температуре 8000С. Затем тигель осторожно вынимают и ставят в эксикатор, который закрывают крышкой через 5 – 10 секунд. После охлаждения в течение 20 – 30 минут в эксикаторе повторно взвешивают на аналитических весах. И так до получения «постоянной массы», то есть когда разница в массе будет не более 0,2 – 0,3 мг. Таким образом, получают массу ”р”.
Запись данных. Данные записывают с точностью ± 0,1 мг по форме:
масса тигеля (пустого):
ое взвешивание т1= 14,8674 г
- ое взвешивание т2 = 14,8670 г
- ое взвешивание т3= 14,8672 г и т.д.
Масса тигеля с сульфатом бария:
ое взвешивание р1= 15,3774 г
- ое взвешивание р2= 15,3726 г
- ъе взвешивание р3=15,3724 г и т.д.
Масса ВаSO4: М = (р3-т3) = 15,3724 – 14,8672=0,5052 г.
Потери при промывании осадка:
П= ν . М. (ПР) 1/2 = 0,2. 233,4. (1,1. 10-10) 1/2=0,0005 г, где = ν – объем промывных вод; М- молярная масса ВаSO4; (ПР)1/2 - растворимость сульфата бария, моль/л.
Расчеты содержания SO42- ведут, используя 4 – 5 значащих цифр по форме:
Х=(М+П) . (М SO4--/МВаSO4) = (М+П) . ФSO4 --/ВаSO4= (М+П). (96,06/233,4)= 0,5057 . 0,04116= 0,02081 г сульфат- иона, где ФSO4-- / ВаSO4 фактор пересчета SO42- по ВаSO4.
Окончательный результат округляют до десятых мг, в данном примере – до 0,0208 г. Получив у преподавателя данные по содержанию SO42-, проводят статистическую обработку результатов анализа по двум результатам (см. раздел «Достоверность анализа»).
Отчет: 1) кратко описывают сущность метода и методику определения; 2) записывают все результаты взвешивания, даже если они совпадают; 3) записывают все расчеты и результаты статистической обработки; 4) делают заключение о результатах анализа.
Техника безопасности: При выполнении анализа необходимо соблюдать следующие меры предосторожности:
- проверить заземление сушильного шкафа и муфельной печи;
- помещать и доставать тигель в шкаф и печь с помощью тигельных щипцов. Нельзя брать тигли пальцами во избежание ожога и загрязнения тигля, а также помещать горячие тигли на дерево или пластикат;
- растворимые соли бария довольно токсичны, поэтому перед приемом пищи и после работы необходимо тщательно мыть руки.
Лабораторная работа№11
Определение содержания никеля
Цель работы: практически освоить количественное определение никеля методом осаждения аморфных осадков органическим осадителем.
Сущность метода: с целью количественного отделения железа от никеля используют метод осаждения раствором аммиака аморфного осадка Fe(OН)3. Из фильтрата никель осаждают диметилглиоксимом.
Аппаратура:
аналитические весы, сушильный шкаф, муфельная печь, насос Камовского или водоструйный насос.
Посуда: воронка, фильтр Шотта, стаканы вместимостью 400 и 600 мл; фильтры «красная лента».
Реактивы: хлорид аммония (твердый); концентрированная и 2 М азотная кислота; 12% раствор аммиака(1:1); 1% аммиачный раствор диметилглиоксима; 2 и 6 М растворы НСI; 0,05 М раствор нитрата серебра.
Методика определения. Предварительно доводят до постоянной массы фильтрующий тигель или воронку Шотта № 40. Температура сушки 1200С, время- 20 минут. Горячий фильтр, чтобы он не треснул, берут подогретыми пинцетом или щипцами и помещают на 1 минуту для охлаждения в деревянный штатив, а затем в эксикатор. После охлаждения в течении 20-30 минут взвешивают на аналитических весах. Операцию повторяют до получения “постоянной массы”, то есть когда разница в массах последующего и предыдущего взвешиваний будет не более 0,2 мг.
Осаждение. Фильтрат после отделения железа (III) или задачу с одним никелем (II), разбавленную 100 мл воды, нагревают почти до кипения, добавляют 6 М раствор НСI до кислой реакции и 10 – 15 (20 мл) мл 1% аммиачного раствора диметилглиоксима (рН=3 – 5). Затем по каплям при постоянном перемешивании добавляют 12% раствор аммиака до его слабого запаха. После отстаивания осадка в течение 20 – 30 минут на водяной бане (осадок иногда всплывает) и охлаждения в течение часа делается проба на полноту осаждения никеля: добавляют 3 – 4 капли 1% раствора диметилглиоксима, если образуется осадок, повторяют осаждение, но меньшей порцией диметилглиоксима.
Фильтрование. Осадок фильтруют через предварительно доведенный до постоянной массы фильтрующий тигель или воронку № 40, используя водоструйный насос или насос Камовского. Сначала через фильтр пропускают всю прозрачную жидкость. Затем в стакане осадок промывают 3 – 4 раза водой порциями по 15 – 20 мл и количественно переносят на фильтр. На фильтре осадок промывают 4 – 6 раз холодной водой порциями по 10 – 15 мл до отрицательной реакции на хлорид-ионы.
Сушка. Осадок сушат в сушильном шкафу в течение 30 – 40 минут при температуре 120 0С, охлаждают сначала в деревянном штативе, затем в эксикаторе, данные по взвешиванию записывают в журнал. Высушивание повторяют в течение 15 – 20 минут до постоянной массы.
Гравиметрическая форма: NiC8 H14 N4O4, фактор пересчета – 0,2031.
Расчеты и форма отчета аналогичны приведенным в предыдущей работе.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Для анализа веществ используют химические реакции, сопровождающиеся изменением физических свойств анализируемой системы, например ее цвета, интенсивности окраски, прозрачности, флуоресценции, электро- и теплопроводности и т.д. Методы анализа, основанные на установлении взаимосвязи между качественным и количественным составом и физическими свойствами исследуемых систем называют физическими, если же процесс определения сопряжен с протеканием химической или электро-химической реакции, то метод называют физико-химическим. Различают прямые и косвенные физико-химические методы анализа. В прямых методах анализа данное (физическое) свойство является критерием содержания определяемого вещества, эти методы основаны на изучении диаграмм «состав-свойство». В косвенных методах физическое свойство системы используют для фиксирования окончания процесса взаимодействия определяемого вещества с реактивом точно известной концентрации. Физико-химические методы отличаются высокой чувствительностью и экспрессностью, позволяют автоматизировать химико-аналитические определения.
Физико-химические методы анализа подразделяют на электрохимические и спектральные (оптические). К электрохимическим методам анализа относят методы, основанные на измерении и регистрации электрических параметров анализируемых систем, например: количества электричества прошедшего через раствор электролита; силы предельного диффузионного тока; электропроводности; потенциала погруженного в исследуемый раствор электрода, изменяющегося в результате протекания химических реакций и т.п. К группе электрохимических методов анализа относят следующие виды анализа: полярографию, кулонометрию, кондуктометрию, потенциометрию и т.д. К спектральным (оптическим) методам анализа относят методы, основанные на изучении эмиссионных и абсорбционных спектров веществ в инфракрасной, видимой, ультрафиолетовой и других областях спектра, а также методы, основанные на измерении интенсивности поглощаемого, излучаемого, отражаемого и рассеиваемого света, например: атомно-абсорбционную спектрофотометрию, абсорбционную спектроскопию, люминесцентную спектроскопию (флуориметрию), пламенную спектрофотометрию, турбидиметрию, нефелометрию и другие.
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Лабораторная работа №1
Градуировка стеклянного ионоселективного электрода и измерение рН прямым потенциометрическим методом
Цель работы. Изучить и освоить теоретические основы прямой потенциометрии, прямого определения рН растворов стеклянным электродом; освоить технику прямой потенциометрии с особенностями градуировки стеклянного электрода с помощью рН-метра
Задание. Проградуировать стеклянный ионоселективный электрод на ионы водорода с погрешностью 0,05 рН в области значений соблюдения линейно-Нернстовской зависимости рН растворов 1,00 – 10,00 (12,00), по двум стандартным буферным растворам рН = 1,68 и рН = 9,18, оценить погрешность измерения рН растворов.
Теоретическое введение. Сущность прямой потенциометрии, теоретические основы потенциометрического измерения концентрации ионов водорода и рН растворов стеклянным ионоселективным электродом на ионы водорода (см. учебную литературу и лекции).
Обоснование методики. Смысл градуировки стеклянного ионоселективного электрода на ионы водорода с помощью рН-метра заключается в приведении прибора в состояние, при котором можно использовать всю линейно-Нернстовскую зависимость изменения потенциала стеклянного электрода от рН.
Для измерения рН с погрешностью 0,05 требуется проградуировать стеклянный электрод с помощью ручек управления рН-метра. Градуировка проводится методом подбора по двум стандартным буферным растворам рНст = 1,68 и рНст = 9,18 ручками «калибровка» и «крутизна». Проградуировать стеклянный электрод удается благодаря тому, что ручка «калибровка» изменяет значения рН как в кислой так и щелочной областях рН на одинаковое значение (рНкал = рНкр. рНкал = 1,00, рН = 1,68 + 1,00 = 2,68 и рН = 9,18 1,00 = 10,18 – вероятность градуировки одной ручкой «калибровка» мала). Ручка «крутизна» меняет значение рН в щелочной области в 2 – 4 раза больше, чем в кислой (рНкал = 1,00; рНкал рНкр. рН = 1,68 + 1,00 = 2,18, рН = 9,18+1,00 3 = 12,18). Наибольший вклад в градуировку вносит ручка «крутизна», поэтому градуировка осуществляется попеременно двумя ручками. Начинают градуировку с кислого раствора (рН = 1,68) и корректируют значения рН ручкой «калибровка», затем меняют кислый раствор на щелочной (рН = 9,18) и корректируют значения рН ручкой «крутизна».
Экспериментальная часть
Аппаратура:
рН-метр любой марки рН-121;
стеклянный ионоселективный электрод;
насыщенный хлоридсеребряный электрод сравнения;
электромагнитная мешалка.
Посуда и реактивы:
стаканчика вместимостью 100 мл или 50 мл – 3 шт,
стандарт-титры для рН-метра с рН = 1,68 и рН = 9,18;
дистиллированная вода.
Выполнение работы:
Подключают прибор к сети 220 В с помощью сетевого шнура и прогревают 15 – 20 минут.
- Ручкой «температура раствора» устанавливают по верхней шкале прибора температуру раствора, рН которого измеряют.
- Индикаторный электрод перед каждым погружением в измеряемый раствор тщательно промывают дистиллированной водой и осторожно удаляют воду с его поверхности фильтровальной бумагой. Эту операцию проводят только при отключенной электродной ячейке (черная кнопка рН-метра нажата).
- Проверку и настройку рН-метра проводят по двум стандартным растворам с рН = 1,68 и рН = 9,18.
- В стаканчик наливают стандартный буферный раствор и опускают в него электроды.
- Нажимают клавишу диапазона измерений – «1 - 14» (грубая шкала), а затем клавишу – «1 – 4» (точная шкала) и устанавливают рН = 1,68 с помощью ручки управления «калибровка», повторяют операцию «3».
- Проверяют прибор по раствору с рН = 9,18. Нажимают клавишу «1 – 14» устанавливают рН – 9,2 ручкой крутизны (или двумя ручками «крутизна» и «калибровка») нажимают клавишу «9 – 14» и устанавливают рН = 9,18 с помощью ручки управления «крутизна».
- Проверяют прибор по раствору с рН = 1,68, и если получают значение 1,68 ± 0,05, то стеклянный электрод проградуирован. Если рН отличается больше, чем на 0,05, цикл градуировки повторяют до нужного интервала рН = 1,68 ± 0,05 и 9,18 ± 0,05.
- По окончании работы прибор отключают от сети, электроды осторожно промывают дистиллированной водой и оставляют погруженными в воду.
Запись результатов:
изм. калибр.
1

 ). рН = 1,68 1,90 1,68
). рН = 1,68 1,90 1,68изм. крут.
 2). рН = 9,18 9,29 9,15 (рН в пределах 0,05 рН)
2). рН = 9,18 9,29 9,15 (рН в пределах 0,05 рН)изм.
 n). РН = 1,68 1,68 0,05
n). РН = 1,68 1,68 0,05