
Отчет о научно-исследовательской работе «Разработка Концепции обеспечения качества лекарственных средств в Российской Федерации»
| Вид материала | Отчет |
- Отчет о научно-исследовательской работе разработка концепции архитектуры программного, 551.78kb.
- Отчет о научно-исследовательской и опытно-конструктороской работе, 3288.39kb.
- Национальный стандарт российской федерации производство лекарственных средств система, 1337.84kb.
- Отчет о научно-исследовательской работе, проведенной по заказу Министерства экономического, 6886.49kb.
- Отчет о научно-исследовательской и опытно-конструкторской работе по теме, 5626.08kb.
- Отчет о научно-исследовательской и опытно-конструкторской работе по теме, 2858.14kb.
- Отчет о научно-исследовательской работе, 392.92kb.
- Отчет о научно-исследовательской работе «разработка концепции развития корпоративного, 4114.59kb.
- Отчет о научно-исследовательской работе разработка концепции Объединенных Государственных, 6757.77kb.
- Реферат отчет о научно-исследовательской работе состоит, 61.67kb.
1.2 Руководства и рекомендации
Руководства, используются государством для уточнения тех или иных требований к уровню качества, объяснения специфических подходов к регулированию отдельных групп препаратов (например, синтетические пептиды или комбинированные препараты) или разъяснения позиции государства при проведении экспертизы представленных заявителем документов (например, оценка данных по стабильности).
Также существуют международные руководства, отражающие процесс гармонизации требований различных государств к качеству лекарственных средств – руководства ВОЗ и Международной конференции по гармонизации (ICH).
1.3 Международная конференция по гармонизации (ICH)
Стремительное развитие международной фармацевтической отрасли в 70-80 гг. 20 века и глобализация фармацевтического рынка стали тормозиться разобщенными национальными системами регистрации лекарственных средств, в первую очередь различиями в технических требованиях. Наряду с этим, рост стоимости затрат на здравоохранение, на научно-исследовательские работы по созданию новых лекарственных средств, необходимость быстрого доступа населения к современным более эффективным препаратам требовали проведения гармонизации регуляторных требований.
В 1989 году на Парижской конференции органов по регулированию лекарственных средств, ежегодно проводимой ВОЗ, этот вопрос начал решаться регуляторными органами США, ЕС и Японии.
В апреле 1990 года представители агентств этих стран и ассоциаций производителей создали Международную конференцию по гармонизации, секретариат которой расположен в Женеве в штаб-квартире Международной федерации фармацевтических производителей ассоциаций. (IFPMA).
Первоначальной задачей ICH была гармонизация технических требований к регистрационному досье, подаваемому в ЕС, США и Японии. По мере успешной работы конференции ее задачи были расширены.
Основные задачи ICH на текущее десятилетие были определены на ее 5-ой конференции в Сан-Диего в 2000 году:
- создание форума для конструктивного диалога между регуляторными органами и фармацевтической промышленностью в части действующих и объективных различий в регистрационных требованиях в США, ЕС и Японии с целью обеспечить более быстрое внедрение в практику новых медицинских продуктов и доступ к ним пациентов;
- участие в защите общественного здоровья с международных перспектив;
- мониторинг и обновление гармонизированных технических требований, ведущих к большему взаимному признанию данных по исследованиям и разработке лекарственных средств;
- исключение в будущем различных требований путем гармонизации выбранных областей, необходимых для дальнейшего развития терапии и новых технологий производства медицинской продукции;
- обеспечение распространения и понимания гармонизированных руководств и походов, которые обновляют или заменяют текущие положения и позволяют более экономно использовать человеческие и материальные ресурсы без ущерба безопасности;
- обеспечение распространения и понимания гармонизированных руководств, их использования для внедрения и объединения общих стандартов.
На сегодняшний день в ICH входит 6 членов, 3 наблюдателя (без права голосования) и IFPMA.
Члены ICH представлены регуляторными органами ЕС, США и Японии и ассоциациями фармацевтических производителей этих стран (регионов), где разрабатывается, производится и продается наибольшее количество лекарственных средств:
- От Европейского Союза в работе ICH принимает участие Европейское агенство по медицинским продуктам (EMEA) и Европейская Федерация фармацевтических производителей и ассоциаций (EFPIA).
- От США в ICH входят Администрация по пищевым продуктам и лекарственным средствам (FDA) США и Ассоциация фармацевтических разработчиков и производителей США (PhRMA).
- От Японии в работе по гармонизации участвуют Агентство по лекарственным средствам и медицинским изделиям Министерства здравоохранения, труда и социальных вопросов Японии и Национальный институт наук в области здоровья, а также Ассоциация японских фармацевтических производителей (JPMA).
Наблюдатели в ICH рассматриваются как посредники со странами и регионами, не входящими в ICH.
В первую очередь, это Всемирная организация здравоохранения, Европейская Ассоциация свободной торговли, представленная Swissmedic Switzerland, и Канада в лице Министерства здравоохранения Канады.
Также работе ICH помогает Международная федерация фармацевтических производителей и ассоциаций, на базе которой работает секретариат ICH.
Деятельность ICH организуется Исполнительным комитетом, в котором каждый из 6 членов имеет по 2 места с правом голоса, а наблюдатели и IFPMA назначают участников комитета без права голоса. Технические функции по организации работы выполняет секретариат ICH. Основным методом разработки руководств является использование экспертных рабочих групп (EWG), рабочих групп по внедрению (IWG) и неформальных рабочих групп, в дальнейшем также предполагается использование видеоконференций и электронных коммуникаций.
На сегодняшний день руководства ICH разделены на 4 основных раздела:
- безопасность (safety)
| Код документа | Название руководства |
| Исследования на мутагенность | |
| S1A | Необходимость в исследовании мутагенности препаратов |
| S1B | Проверка на мутагенность препаратов |
| S1C(R1) | Отбор доз для исследований мутагенности препаратов и дозовый предел |
| S2A | Руководство по специфическим аспектам испытаний регулятивной генотоксичности для препараов |
| S2B | Генотоксичность: A Standard Battery for Genotoxicity Testing of Pharmaceuticals |
| S3A | Примечание к руководству Токсикокинетики: Оценка общей экспозиции в исследованиях токсичности |
| S3B | Фармакокинетика: Руководство по исследованию распределения повторной дозы в тканях |
| Проверка на токсичность | |
| S4 | Испытания токсичности однократной дозы |
| S4A | Продолжительность испытаний постоянной токсичности на животных (Испытание токсичности на грызунах и не грызунах) |
| Генеративная токсикология | |
| S5(R2) | Обнаружение токсичности медицинских продуктов для репродуктивности и токсичность к воспроизведению потомства у мужчин |
| S5A | Поддержка ICH Нормативы токсичности для мужской плодовитости |
| Биотехнологические продукты | |
| S6 | Оценка доклинической безопасности биотехнологически полученных лекарств |
| Исследования фармакологии | |
| S7A | Исследования фармакологии безопасности для лекарств людей |
| S7B | Неклиническая оценка потенциала для запаздывающей желудочковой реполяризации (QT промежуточная пролонгация) лекарств для людей |
| Иммунотоксикологические исследования | |
| S8 | Иммунотоксикологические исследования на лекарствах для людей |
- эффективность (efficacy)
| Безопасность клинических исследований | |
| E1 | Количество пациентов, которые подвергаются клиническому изучению безопасности лекарств, предназначенных для длительного лечения состояний не угрожающих жизни |
| E2A | Управление данными по клинической безопасности: определения и стандарты для срочного отчета |
| E2B(R3) | Управление данными по клинической безопасности: элемент данных для переноса сообщений безопасности особого случая |
| E2C(R1) | Управление данными клинической безопасности: Периодическое обновление отчетности по безопасности для продаваемых лекарств Приложение к E2C: Периодическое обновление отчетности по безопасности для продаваемых лекарств в E2C(R1)) |
| E2D | Управление данными о безопасности после вывода на рынок: Определения и стандарты для отчетов |
| E2E | Планирование фармаконадзора |
| Отчеты по клиническим исследованиям | |
| E3 | Структура и содержание Отчетов по клиническим исследованиям |
| Исследования эффекта в зависимости от дозы | |
| E4 | Доза-эффект информация для внесения данных в досье по регистрации |
| Этнические факторы | |
| E5(R1) | Этнические факторы в приемлемости иностранных клинических данных |
| GCP (Надлежащая клиническая практика) | |
| E6(R1) | GCP (Надлежащая клиническая практика) |
| Клинические испытания | |
| E7 | Исследования в подтверждение по специфическим популяциям: гериатрия |
| E8 | Основное рассмотрение клинических испытаний |
| E9 | Статистические принципы для клинических испытаний |
| E10 | Выбор контрольной группы и связанных данных в клинических испытаниях |
| E11 | Клиническое исследование медицинских продуктов на детей |
| Нормативы клинической оценки по терапевтической категории | |
| E12 | Принципы клинической оценки новых антигипертензивных препаратов |
| Клиническая оценка | |
| E14 | Клиническая оценка QT/QTc интервала пролонгации и проаритмического потенциала для неантиаритмических препаратов |
| Фармакогеномика | |
| E15 | Терминология в фармакогеномике |
- качество (quality)
Перечень документов ICH в разделе «Качество»
| Код документа | Название руководства |
| Стабильность | |
| Q1A(R2) | Stability Testing of New Drug Substances and Products «Испытание стабильности новых фармацевтических субстанций и препаратов» |
| Q1B | Stability Testing : Photostability Testing of New Drug Substances and Products «Испытание фотостабильности новых фармацевтических субстанций и препаратов» |
| Q1C | Stability Testing for New Dosage Forms «Испытание стабильности новых лекарственных форм» |
| Q1D | Bracketing and Matrixing Designs for Stability Testing of New Drug Substances and Products «Методы группировки для проведения испытания стабильности новых фармацевтических субстанций и препаратов» |
| Q1E | Evaluation of Stability Data «Оценка данных по стабильности» |
| Q1F | Stability Data Package for Registration Applications in Climatic Zones III and IV «Объем данных по стабильности для регистрационных досье на препараты, использующиеся в климатических зонах III и IV» |
| Валидация | |
| Q2(R1) | New title: Validation of Analytical Procedures: Text and Methodology Previously: Text on Validation of Analytical Procedures Новое название: «Валидация аналитических методик: Содержание и методология» взамен руководств «Содержание валидации аналитических методик» и «Валидация аналитических методик: методология» |
| Примеси | |
| Q3A(R2) | Impurities in New Drug Substances «Примеси в новых фармацевтических субстанциях» |
| Q3B(R2) | Impurities in New Drug Products «Примеси в новых лекарственных препаратах» |
| Q3C(R2) | Impurities: Guideline for Residual Solvents «Примеси: руководство по остаточным растворителям» |
| Фармакопея | |
| Q4 | Pharmacopoeias «Фармакопеи» |
| Q4A | Pharmacopoeial Harmonisation «Гармонизация фармакопей» |
| Q4B | Regulatory Acceptance of Analytical Procedures and/or Acceptance Criteria (RAAPAC) «Признание регуляторными органами аналитических методик и/или критериев приемлемости» |
| Качество биотехнологических препаратов | |
| Q5A(R1) | Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin «Оценка вирусной безопасности биотехнологических продуктов, полученных из штаммов клеток человека и животных» |
| Q5B | Quality of Biotechnological Products : Analysis of the Expression Construct in Cells Used for Production of r-DNA Derived Protein Products «Качество биотехнологических препаратов: анализ экспрессирующих генных конструкций в клетках, использующихся для получения протеиновых препаратов с использованием рекомбинантной ДНК» |
| Q5C | Quality of Biotechnological Products : Stability Testing of Biotechnological/Biological Products «Качество биотехнологических препаратов; оценка стабильности биотехнологических/биологических препаратов» |
| Q5D | Derivation and Characterisation of Cell Substrates Used for Production of Biotechnological/Biological Products «Получение и характеристики клеточных субстратов, используемых в производстве биотехнологических/биологических препаратов» |
| Q5E | Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process «Сравнимость (идентичность) биотехнологических/биологических препаратов в случае изменений в технологическом процессе их получения» |
| Спецификации | |
| Q6A | Specifications : Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances (including Decision Trees) «Спецификации: Параметры качества и критерии приемлемости для новых фармацевтических субстанций и лекарственных препаратов: химические субстанции (включая алгоритмы)» |
| Q6B | Specifications : Test Procedures and Acceptance Criteria for Biotechnological/Biological Products «Спецификации: Параметры качества и критерии приемлемости для биотехнологических/биологических препаратов» |
| Надлежащая производственная практика | |
| Q7 | Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients «Руководство по надлежащей производственной практике для активных фармацевтических компонентов» |
| Разработка фармацевтических продуктов | |
| Q8 | Pharmaceutical Development «Разработка фармацевтических продуктов» |
| Управление рисками, связанными с качеством | |
| Q9 | Quality Risk Management «Управление рисками, связанными с качеством» |
| Q10 | Pharmaceutical Quality system «Система качества на фармацевтическом предприятии» Стадия 3. |
- междисциплинарные документы (m)
| M1 | Медицинская терминология |
| M2 | Электронные образцы для переноса регулятивной информации |
| M3(R1) | Синхронизация доклинических исследований в отношении к клиническим испытаниям |
| M4 | Общий технический документ (ОТД) |
| M5 | Элементы данных и образцов для Лекарственных словарей |
Первым руководством ICH, гармонизировавшим формат представляемых данных, стало руководство Е3 «Содержание и формат отчетов о клинических исследованиях».
Все работы ICH делятся на 4 категории:
- Формальные процедуры ICH по разработке новых руководств
- Процедуры по созданию перечня «Вопросов и ответов» для облегчения использования действующих руководств
- Процедуры пересмотра или внесения изменений в действующие руководства
- Процедуры актуализации, т.е. добавления стандартов в существующие руководства и/или рекомендации.
Разработка нового документа ICH начинается с подачи предложения о разработке руководства. Несмотря на то, что предложения о разработке могут поступать из различных источников, формальное предложение о разработке должен член или наблюдатель ICH. В предложении (концептуальная записка) указывается тип предлагаемой работы, состояние обсуждаемой проблемы, краткое описание технических и научных вопросов, требующих гармонизации, дополнительная информация, тип экспертной рабочей группы. Исполнительный комитет рассматривает предложение и принимает решение о принятии или непринятии новой темы. Для решения может потребоваться переработка предложения и/или разработка плана работ по установленному ICH образцу. В зависимости от типа предлагаемых работ Исполнительный комитет может организовать экспертную рабочую группу (EWG) или рабочую группу по внедрению (IWG), которой может быть поручено доработать предложение о разработке и/или план разработки. Рабочая группа формируется из 2-х официально назначенных представителей от каждого из 6 членов ICH, и по одному представителю могут предложить наблюдатели и заинтересованные стороны, если это применимо.
Рисунок 1 иллюстрирует процесс разработки новых руководств, их согласования и введения в действие.

Рис. 1 процесс разработки новых руководств
Как уже упоминалось ранее, для улучшения практического внедрения руководства обычно разрабатываются рекомендации в виде перечня вопросов и ответов. При разработке такого типа документов собираются вопросы (и поступающие на сервер ICH, и получаемые членами ICH) и направляются в рабочую группу по внедрению (IWG). Они обрабатываются, переформулируются и вместе с предлагаемыми ответами представляются Исполнительному комитету. Затем документ проходит этап 2 и этап 4 по схеме, описанной ранее.
Если принятое руководство нуждается в пересмотре, то используется вышеописанная процедура принятия руководства, стоящая из 5 этапов. Единственное различие – окончательный документ будет представлять собой пересмотренную версию уже действующего документа с дополнительным индексом R1, R2 и т.д.
При необходимости актуализации руководства, например в связи с появлением новых данных, процесс также будет включать оценку данных экспертной рабочей группой, обсуждение и согласование членами ICH.
1.4 Обеспечение качества лекарственного средства
Обеспечение качества лекарственного средства – это широкое понятие, охватывающее все факторы, которые по отдельности или вместе влияют на качество продукции.
Создание системы качества, предназначенной для гарантии безопасности и эффективности препарата, основанной на научных исследованиях и анализе рисков, на каждой стадии жизненного цикла, будет способствовать качеству и его улучшению в течение всего жизненного цикла продукта.
Жизненный цикл продукта включает следующие виды деятельности для новых и существующих лекарственных средств:
- Фармацевтическая разработка
- Опытно-промышленная проверка и перенос технологии
- Промышленное (серийное) производство
- Продажа и применение препарата
- Прекращение выпуска продукта
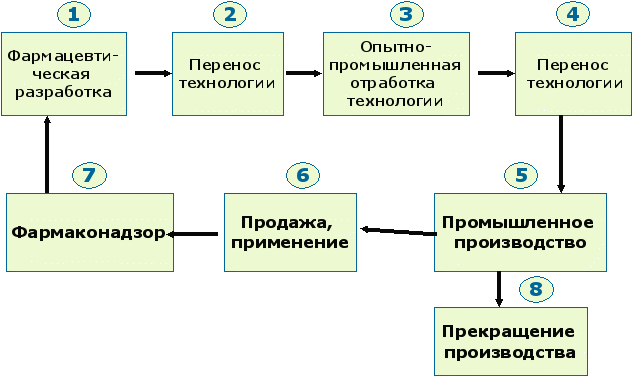
Рис.2 Обеспечение качества на всех этапах жизненного цикла лекарства
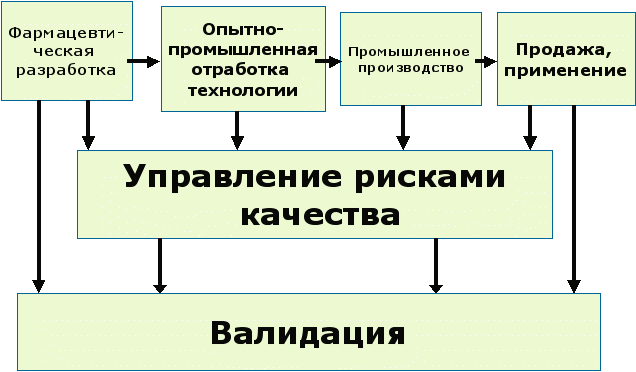
Рис.3 Управление рисками и валидация на этапах жизненного цикла лекарства
Управление рисками качества является неотъемлемой составной частью эффективной системы фармацевтического качества. Оно может предоставлять действенный подход для идентификации, научно обоснованной оценки и управления потенциальными рисками в отношении качества. Оно облегчает постоянное улучшение эффективности процесса и качества продукции в течение жизненного цикла продукта.
Инструменты теории рисков:
• Элементарные методики помощи в управлении рисками (сетевые графики, контрольные карты и т.д.);
• Анализ характера и последствий отказа (FMEA);
• Анализ характера, последствий и критичности отказа (FMECA);
• Анализ дерева ошибок (FTA);
• Анализ опасности и критические контрольные точки (HACCP);
• Исследования опасности и пригодности к эксплуатации (HAZOP);
• Предварительный анализ опасности (PHA);
• Ранжирование и фильтрация рисков;
• Поддерживающие статистические инструменты
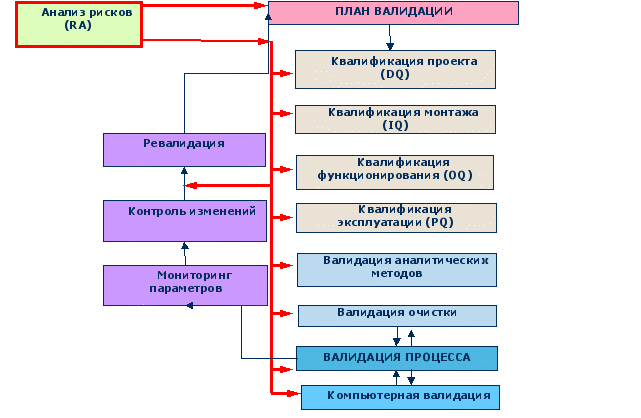
Рис.4 Алгоритм валидации
Документы валидации:
- валидационный план;
- валидационный протокол;
- отчет о проведении валидации;
- документация, прилагаемая к отчету (применявшиеся инструкции, первичные данные измерений, распечатки, отчеты о калибровке, диаграммы расстановки оборудования и схемы этапов работ, полные аналитические и измерительные протоколы).
1.4.1 Цель фармацевтической разработки
Создать продукт высокого качества и соответствующий производственный процесс, обеспечивающий клиническое действие продукта, предусмотренное его дизайном.
Фармацевтическая разработка включает:
• Определение параметров качества целевого продукта
• Идентификацию критических свойств качества препарата
• Влияние вспомогательных веществ
• Разработку технологического процесса
- Идентификацию и оценку рисков, свойств материалов и параметров процесса, которые могут иметь влияние на критические свойства продукта
- Определение функциональных отношений, связывающих свойства материала и параметры процесса с критическими свойствами продукта
- Определение функциональных отношений, связывающих свойства материала и параметры процесса с критическими свойствами продукта
- Использование понимания продукта и процесса в комбинации с управлением рисками качества.
Сведения о лекарственной субстанции
- физико-химические свойства,
- биологические свойства,
- содержание влаги,
- размер частиц,
- свойство кристаллов,
- биологическая активность,
- способность проникать сквозь биологические мембраны.
Вспомогательные вещества
- Концентрация, характеристики, влияющие на стабильность, биодоступность, технологичность;
- Функциональные возможности антиоксидантов, веществ, увеличивающих всасывание, способствующие распадаемости и высвобождению.
Технологический процесс
Параметры технологического процесса, их влияние на технологичность и качество продукта, функционирование процесса в различных режимах, масштабах с использованием различного оборудования.
Пространство дизайна
Многофакторное сочетание и взаимодействие входящих переменных (свойства материалов) и параметров процессов способных обеспечить качество продукта.
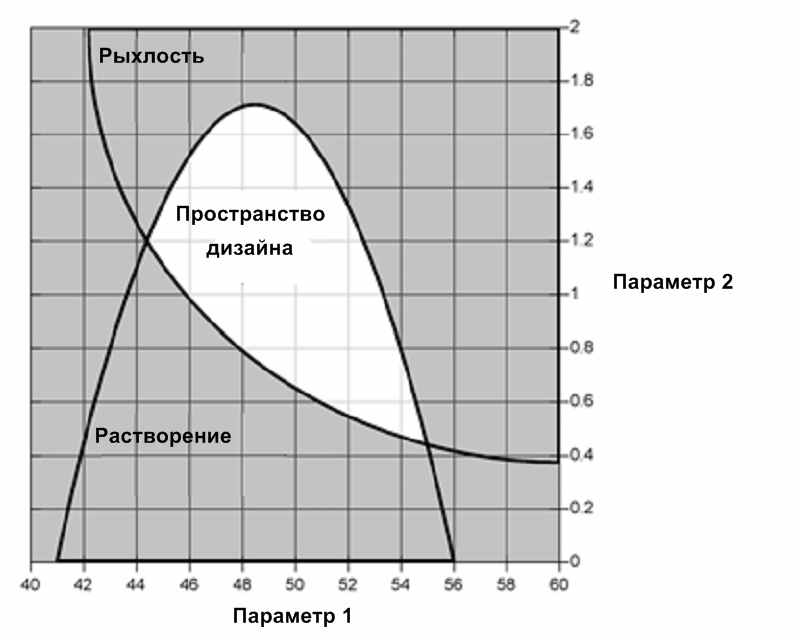
Рис. 5: Предложенное пространство дизайна, включающее пересечение областей диапазонов рыхлости и растворения таблетки
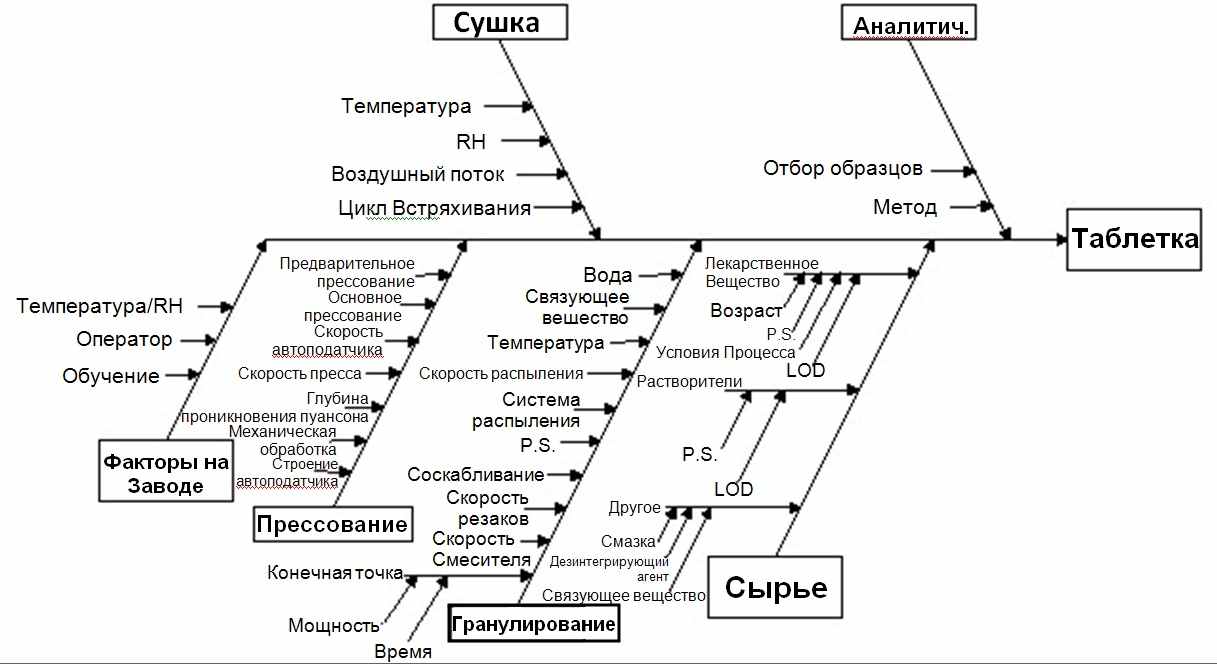
Рис.6 Диаграмма Ишихавы для определения рисков в производстве таблеток

Рис.7 Структура этапов фармацевтической разработки
Досье на регистрацию
В соответствии с Общим техническим документом –ICH (М4) структура досье на регистрацию в США, ЕС и Японии идентична (сравнение с российскими требованиями к досье на регистрацию дано во второй главе).
1.4.2. Опытно-промышленная проверка
- Опытно-промышленная проверка позволяет разработать исходные данные на проектирование и опытно-промышленный регламент;
- Провести масштабирование технологического процесса;
- Обеспечить перенос зарегистрированного продукта.
Перенос технологии
Целью деятельности по переносу технологии является передача знаний о продукте и процессе между стадией разработки и стадией производства, и внутри или между производственными участками. Эти знания образуют основу технологического процесса, стратегии управления и контроля, подходов к валидации процесса.
Надлежащая инженерная практика(GEP)
Проверенные инженерные методы и стандарты, которые применяются в течение создания и эксплуатации производства с целью получения адекватных и эффективных по цене решений.
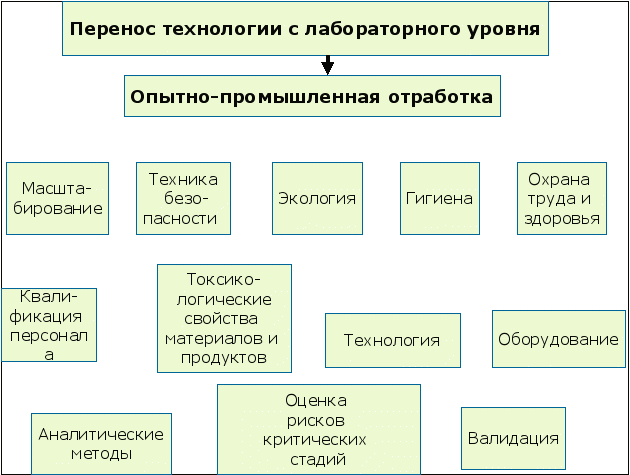
Рис. 8 Структура документов, получаемых на этапе опытно-промышленной отработки
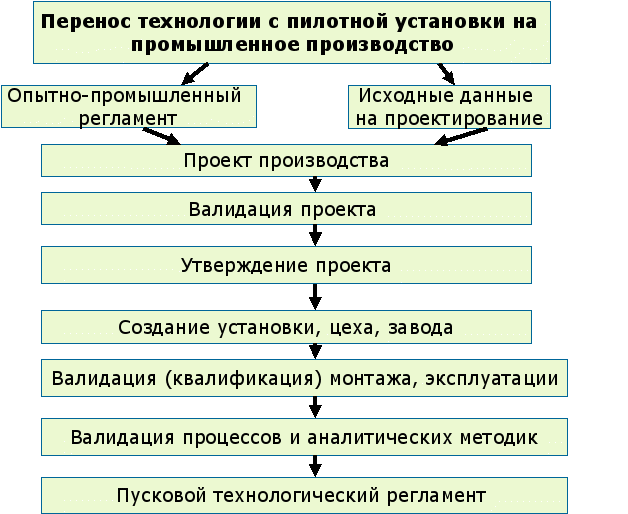
Рис. 9 Перенос технологии в промышленное производство
1.4.3. Промышленное производство
Цели производственной деятельности включают достижение концепции заданного продукта, обоснование и поддержание контролируемого состояния и содействие постоянному улучшению. Система фармацевтического качества должна обеспечивать постоянное достижение заданного уровня качества продукта, подходящих параметров процесса, соответствующего набора методов контроля, идентификацию и оценку возможностей улучшения и постоянное расширение базы имеющихся знаний.
При производстве лекарственного средства обязательным является:
- Разработка и утверждение пускового или промышленного регламента;
- Разработка локальных актов (документов) системы обеспечения качества производства;
- Обучение по GMP;
- Валидация;
- Закупка и контроль материалов;
- Система хранения сырья и материалов;
- Система карантина;
- Контроль и обеспечение качества;
- Выпуск на реализацию;
- Годовой отчет по качеству;
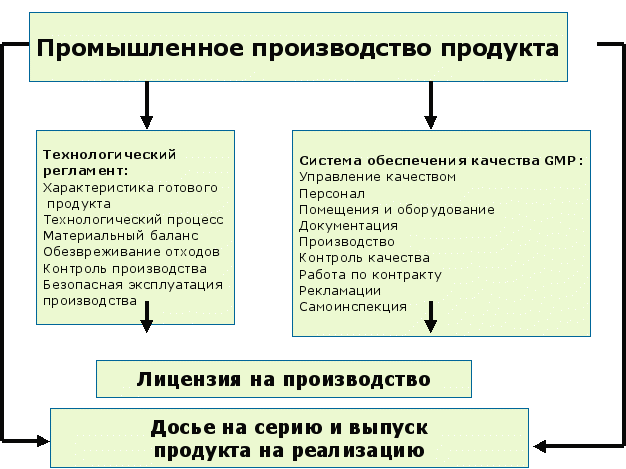
Рис. 10 Основные факторы, обеспечивающие промышленное производство
1.4.4.Прекращение выпуска продукта должно обеспечить:
- сохранение документации;
- сохранение образцов;
- продолжение оценки качества продукта и оформление отчетов;
- продолжение изучения стабильности.
Продажа лекарственного средства руководствуется следующими системами:
- Система оптовой продажи – GDP.
- Система розничной продажи – GPP.
- Система контроля качества и фармаконадзор.
1.4.5.Факторы концепции обеспечения качества
- Концепция, организация и документация системы фармацевтического качества должны быть хорошо структурированными и ясными для того чтобы облегчить общее понимание и последовательное применение.
- Элементы концепции должны применяться таким образом, чтобы они были подходящими и соразмерными для каждой из стадий жизненного цикла продукта, с учетом разницы между ними и различных целей на каждой стадии.
- Система фармацевтического качества должна включать следующие элементы:
- мониторинг параметров процесса и качества продукта,
- корректирующие и предупреждающие действия,
- управление изменениями и анализ со стороны руководства.
- Должны быть установлены и использованы индикаторы функционирования для мониторинга эффективности процессов в рамках системы фармацевтического качества.
Реализация качества на этапах жизненного цикла
| Аспект | Качество по дизайну |
| Фармацевтическая разработка | Систематическое, связывающее механистическое понимание свойств материала и параметров процесса с качеством лекарственного продукта. Проведение многофакторных экспериментов для того, чтобы понять продукт и процесс. Создание пространства дизайна. |
| Производство | Корректируется в пределах пространства дизайна. Подход к валидации учитывает жизненный цикл и, в идеале, непрерывную верификацию процесса. Сосредоточен на стратегии контроля и надежности. Использование статистических методов контроля процесса. |
| Контроль процесса | Используются инструменты PAT с соответствующими методами предшествующего и последующего контроля. Операции процесса даже после утверждения отслеживаются и имеют тенденцию к последующему непрерывному совершенствованию. |
| Спецификации Продукта | Часть общей стратегии контроля качества. Основаны на требуемых характеристиках продукта с соответствующими опорными данными. |
| Контроль качества продукта | Качество лекарственного продукта гарантируется стратегией контроля, основанной на риске, для хорошо изученного продукта и процесса. Контроль качества сдвинут на восходящий поток, с возможностью тестирования выпуска в реальном времени или уменьшенным тестированием конечного продукта. |
1.4.6.Элементы системы фармацевтического качества
- Система мониторинга параметров процессов и качества продуктов.
- Система корректирующих и предупреждающих действий (CAPA).
- Система управления изменениями.
- Анализ со стороны руководства в отношении параметров процессов и качества продуктов.
- Эти элементы должны применяться таким образом, чтобы они были соразмерными и подходящими к каждой стадии жизненного цикла продукта с учетом различий между ними и различных целей на каждой стадии. В течение жизненного цикла продукта компании должны оценивать возможности инновационных подходов для улучшения качества продукта.
- Каждый элемент сопровождается таблицей с примерами его применения на конкретной стадии фармацевтического жизненного цикла.
Система мониторинга параметров процессов и качества продуктов
Фармацевтические компании должны планировать и разрабатывать систему для мониторинга параметров процессов и качества продуктов, чтобы обеспечить поддержание контролируемого состояния. Эффективная система мониторинга дает гарантию постоянной способности процесса и методов контроля производить продукт желаемого качества, и определяют области для постоянного улучшения.
Система мониторинга параметров процессов и качества продуктов должна:
- Использовать управление рисками качества для установления стратегии контроля, которая может включать параметры и атрибуты, относящиеся к материалам и компонентам активных фармацевтических субстанций и готовых лекарственных средств, условиям функционирования помещений и оборудования, внутрипроизводственному контролю, спецификациям готового продукта и к связанным с ними методам и частоте мониторинга и контроля. Стратегия контроля должна облегчить своевременную прямую и обратную связь с потребителями и поставщиками и соответствующие корректирующие и предупреждающие мероприятия.
- Предоставить инструменты для измерения и анализа параметров и атрибутов, определенных в стратегии контроля (например, управление данными и статистические методы).
- Анализировать параметры и атрибуты, определенные в стратегии контроля для проверки непрерывного функционирования в пределах контролируемого состояния.
- Определить источники отклонений, влияющие на параметры процесса и качество продукта, для действий по возможному постоянному улучшению в целях снижения или управления отклонениями.
- Включать обратную связь с потребителями в отношении качества продукта, как из внутренних, так и из внешних источников, например, рекламации, отбраковка продукта, несоответствия, отзывы, отклонения, аудиты, инспекции регуляторных органов и заключения по ним.
- Предоставлять сведения для расширения понимания процесса, обогащения области разработки (там, где это применимо) и способствовать использованию инновационных подходов при валидации процессов.
Применение системы мониторинга параметров процесса и качества продукта в течение жизненного цикла продукта
-
Фармацевти-ческая разработка
Перенос технологии
Серийное производство
Прекращение выпуска продукта
Сведения, полученные о продукте и процессе и их мониторинг, проводимые в течение стадии разработки, могут быть использованы для установления стратегии управления производством
Мониторинг процессов масштабирования может предоставить предварительные показатели параметров процесса и успешную интеграцию в производство. Сведения, полученные в процессе переноса и масштабирования, могут быть использованы в дальнейшем развитии стратегии управления.
Должна применяться хорошо проработанная система для мониторинга параметров процесса и качества продукта, чтобы гарантировать производительность в пределах контролируемого состояния и идентифицировать области для улучшения.
После прекращения производства должен продолжаться мониторинг в объеме контроля стабильности вплоть до завершения этих исследований. Должны продолжаться необходимые действия в отношении выпущенного продукта в соответствии с национальными регуляторными требованиями.
Система корректирующих и предупреждающих действий (САРА)
Фармацевтическая компания должна иметь систему для проведения корректирующих и предупреждающих действий, вытекающих из расследования рекламаций, отбраковки продукта, несоответствий, отзывов, отклонений, аудитов, регуляторных инспекций и наблюдений и тренд-анализов мониторинга параметров процесса и качества продукта. В процессе расследования должен применяться структурный подход с целью определения исходных причин. Уровень усилий и степень формализации расследования должны быть соразмерными уровню риска согласно ICH Q9. Методология САРА должна приводить к улучшениям продукта и процесса и расширению понимания продукта и процесса.
Применение системы корректирующих и предупреждающих действий в течение жизненного цикла продукта
| Фармацевтическая разработка | Перенос технологии | Серийное производство | Прекращение выпуска продукта |
| Изучается вариабельность продукта или процесса. Методология САРА может использоваться там, где корректирующие и предупреждающие действия встроены в повторяющийся процесс проектирования и разработки. | САРА может использоваться как эффективная система для прямой и обратной связи с поставщиками и потребителями и для постоянного улучшения. | Должна использоваться САРА и оцениваться эффективность действий. | Использование САРА должно продолжаться после прекращения выпуска продукта. Должно оцениваться влияние на продукт, остающийся на рынке, а также на другие продукты, которые это может затронуть. |
Система управления изменениями
Инновация, постоянное улучшение, результаты мониторинга параметров процесса и качества продукта и САРА побуждают изменения. Для того чтобы оценивать, санкционировать и внедрять эти изменения надлежащим образом, компания должна иметь эффективную систему управления изменениями. В общем случае имеется разница в формальном подходе к процессам управления изменениями до первоначального представления документов в регуляторные органы и после представления, когда могут потребоваться изменения регуляторной заявки в соответствии с национальными требованиями.
Система управления изменениями обеспечивает своевременное и эффективное проведение постоянного улучшения. Она должна предоставить высокую степень уверенности, что не появятся непреднамеренные последствия внедренного изменения.
Система управления изменениями должна включать следующие моменты в соответствии со стадиями жизненного цикла продукта:
- Для оценки предлагаемых изменений должно использоваться управление рисками качества. Уровень усилий и формальности оценки должен быть соизмеримым с уровнем риска.
- Все изменения должны быть надлежащим образом оценены. Предлагаемые изменения должны оцениваться в отношении разрешения на выпуск продукта в продажу (регистрационного удостоверения), включая пространство проекта (дизайна), там где оно установлено, и/или понимание текущего продукта и процесса. Должна проводиться оценка для определения того, требуется ли изменение документов регистрационного досье в соответствии с национальными требованиями. Как указывается в документе ICH Q8, рабочее движение в пределах пространства проекта не рассматривается как изменение (с позиций перспективы новой подачи документов для регистрации лекарственного средства). Однако, с точки зрения системы фармацевтического качества, все изменения должны оцениваться системой управления изменениями внутри компании.
- Предлагаемые изменения должны оцениваться командой экспертов, предоставляющих надлежащие знания и компетенции из соответствующих областей, например, фармацевтической разработки, производства, качества, регуляторных и медицинских вопросов, и для гарантии того, что изменения технически обоснованы. Должны быть заранее установлены критерии оценки предполагаемых изменений.
- После введения должна быть проведена оценка изменения для подтверждения того, что были достигнуты цели изменения и что нет опасного влияния на качество продукта.
Применение системы управления изменениями в течение жизненного цикла продукта
| Фармацевтическая разработка | Перенос технологии | Серийное производство | Прекращение выпуска продукта |
| Изменение является неотъемлемой частью процесса разработки и должно быть документировано формальность процесса управления изменениями должна согласовываться со стадией фармацевтической разработки. | Система управления изменениями должна обеспечивать управление и документирование корректировок, сделанных в отношении процесса во время переноса технологии. | При коммерческом производстве должна быть в наличии формальная система управления изменениями. Надзор со стороны службы качества должен давать гарантию оценки, основанной на научном подходе и анализе рисков | Любые изменения после прекращения выпуска продукта должны проходить через надлежащую систему управления изменениями. |
Анализ со стороны руководства параметров процесса и качества продукта
Анализ со стороны руководства должен давать гарантию того, что управление параметрами процесса и качеством продукта осуществляется в течение всего жизненного цикла. В зависимости от размера и сложности компании анализ со стороны руководства может представлять собой серию обзоров на различных уровнях менеджмента и должен включать своевременный и эффективный процесс обсуждения и продвижения вопросов о надлежащем качестве до рассмотрения на уровне высшего руководства.
Система анализа со стороны руководства должна включать:
(1) Результаты регуляторных инспекций и замечаний, аудитов и других оценок и обязательств, взятых на себя по отношению к регуляторным органам.
(2) Периодические обзоры качества, которые могут включать:
- Измерения удовлетворенности потребителей, как, например, рекламации со стороны клиентов и отзывы продукта;
- Заключения мониторинга параметров процесса и качества продукта;
- Эффективность изменений процесса и продукта, включая также изменения вследствие корректирующих и предупреждающих действий.
(3) Любые последующие действия, исходящие из предыдущего анализа со стороны руководства.
Система анализа со стороны руководства должна идентифицировать соответствующие действия, такие как:
(1) Улучшения технологических процессов и продуктов.
(2) Предоставление и/или перегруппировка ресурсов, обучение персонала.
(3) Получение и распространение знаний.
Применение анализа со стороны руководства в отношении параметров процесса и качества продукта в течение жизненного цикла продукта
| Фармацевтическая разработка | Перенос технологии | Серийное производство | Прекращение выпуска продукта |
| Могут быть выполнены некоторые аспекты анализа со стороны руководства для гарантии адекватности разработки продукта и процесса. | Должны быть выполнены аспекты анализа со стороны руководства для обеспечения того, что разработанный продукт и процесс можно производить в промышленном масштабе. | Анализ со стороны руководства должен представлять собой структурированную систему, как описано выше, и должен поддерживать постоянное улучшение. | Анализ со стороны руководства должен включать такие пункты, как стабильность продукта и рекламации на продукт. |
Постоянное улучшение системы фармацевтического качества
Этот раздел описывает действия, которые должны выполняться для управления и постоянного улучшения системы фармацевтического качества.
- Анализ системы фармацевтического качества со стороны руководства.
- Мониторинг внутренних и внешних факторов, влияющих на систему фармацевтического качества.
- Результаты анализа и мониторинга со стороны руководства.
Анализ системы фармацевтического качества со стороны руководства
- Измерение достижения целей системы фармацевтического качества.
- Оценка индикаторов функционирования, которая может использоваться для наблюдения за эффективностью процессов, входящих в систему фармацевтического качества, таких как:
(1) жалобы, отклонения, САРА и процессы управления изменениями;
(2) обратная связь с аутсорсинговой деятельностью;
(3) процессы самооценки, включая оценку риска, анализ тенденций и аудиты;
(4) внешние оценки, такие как инспекции и заключения регуляторных органов и аудиты со стороны клиентов.
Мониторинг внутренних и внешних факторов, влияющих на систему фармацевтического качества
Факторы, входящие в мониторинг со стороны руководства, могут включать:
- Появляющиеся регуляторные предписания, руководства и публикации в отношении качества, которые могут влиять на систему фармацевтического качества.
- Инновации, которые могут расширять систему фармацевтического качества.
- Изменения стратегий и целей бизнеса.
- Смена владельца продукта.
Результаты анализа и мониторинга со стороны руководства
Результат анализа системы фармацевтического качества и мониторинга внутренних и внешних факторов со стороны руководства может включать:
- Усовершенствования системы фармацевтического качества и связанных процессов.
- Распределение или перераспределение ресурсов и/или обучение персонала.
- Пересмотр политики качества и целей качества.
- Документирование и своевременное и эффективное обсуждение результатов анализа со стороны руководства и действия, включая доведение соответствующих проблем до сведения высшего руководства.
1.5 Мировой опыт регистрации фармацевтических препаратов
1.5.1 Общие соображения
Регистрация фармацевтических препаратов является центральным звеном государственного регулирования лекарственного рынка по всем параметрам: номенклатура допущенных к продаже средств, их эффективность, безопасность и фармацевтические аспекты качества, информация для врачей и потребителей, условия реализации, во многих странах – также цены. В итоговом докладе Конференции международных экспертов по рациональному использованию лекарств (Найроби, Кения, 25-29 ноября 1985 г.), следующим образом была сформулирована одна из первоочередных обязанностей всех правительств: «создание или укрепление органов нормативного контроля лекарств в целях обеспечения адекватной регистрации лекарств приемлемого качества и безопасности».
По данным ВОЗ, из 192 государств-членов этой организации лишь около одной пятой части (35-40 стран) имеет достаточно эффективную контрольно-разрешительную систему в сфере обращения фармацевтических препаратов. Около одной трети, т.е. примерно 55-60 государств практически не располагают какими-либо возможностями в данной области. В остальных 90-100 странах существующие механизмы государственного регулирования лекарственного рынка недостаточно эффективны.
Современная регистрационная система носит комплексный и профилактический характер. Ее цель: на основании представленных заявителем (спонсором нового препарата) материалов оценить соотношение польза/риск по этому препарату для решения вопроса о его допуске в продажу. Если говорить о фармацевтических аспектах качества препаратов, регистрация (вместе с порядком лицензирования производителей) направлена на определение надежности процессов их производства, а не на проверку качества отдельных образцов. Последняя функция является обязанностью, прежде всего, самого производителя.
Именно по этой причине фармацевтический анализ представленных на регистрацию образцов в индустриальных странах не играет существенной роли. Исключение могут составлять образцы, отобранные в ходе предрегистрационных обследований производителя. В этой связи необходимо напомнить, что последующий государственный контроль качества в мировой практике рассматривается не как самостоятельное направление работы, но как форма проверки соблюдения условий, на которых препарат был зарегистрирован.
Эффективная система регистрации препаратов тесно связана с правилами GMP. Вся технологическая и контрольная документация на производстве должна соответствовать материалам, приложенным к заявкам на регистрацию и одобренным регистрационным органом. Следует ожидать, что в дальнейшем эта связь будет еще более укрепляться. В то же время система регистрации полностью отделена от фармакопейной программы в том смысле, что члены фармакопейных комиссий и комитетов не имеют доступа к регистрационным материалам и не участвуют в процедуре регистрации, кроме России.
В странах, имеющих эффективную контрольно-разрешительную систему (это страны т.н. «золотого миллиарда»), регуляторный механизм в целом, в т.ч. и порядок регистрации, основан на единых подходах и принципах. Ниже рассматриваются следующие основные аспекты современной регистрационной системы:
- правовая основа,
- регистрационная политика,
- виды и формат заявок на регистрацию,
- инструктивно-методические указания по подготовке регистрационных материалов,
- процедура регистрации,
- пост-регистрационные изменения,
- регистрационный орган.
1.5.2 Национальные и региональные особенности регистрации лекарственных средств
Вместе с тем в деталях порядка регистрации имеются национальные и региональные различия. В этом плане можно выделить несколько моделей, в частности американскую (США), европейскую (Евросоюз) и независимую (Канада, Австралия, Швейцария, Норвегия и т.п.). Иногда выделяют, кроме того, японскую модель, однако в настоящее время это не целесообразно, поскольку в Японии проводится глубокая реорганизация регуляторных функций в данной области. По имеющимся данным, эта реорганизация направлена в сторону сближения с европейской и американской моделями на основе материалов ICH.
Национальные и региональные особенности регистрации лекарственных средств отражаются прежде всего в терминологии. В одних странах этот процесс именуется лицензированием фармацевтических продуктов [product licensing], в других выдачей разрешений на сбыт или продажу [Marketing Authorization]. В США процедура допуска новых препаратов в обращение именуется одобрением [approval].
Европейский вариант регистрационной системы отличает сочетание союзных (региональных) и национальных процедур. В рамках ЕС гармонизированы формат и требования к заявке на регистрацию (досье). Входящее в Еврокомиссию Агентство по оценке медикаментов (EMEA) рассматривает т.н. «централизованные заявки» на инновационные и высокотехнологичные препараты и координирует общие подходы к процедуре регистрации. Решения Агентства обязательны для всех стран ЕС. Вместе с тем основной объем работы по регистрации, в частности в отношении дженериков, выполняют национальные регуляторные органы. Их решения, как правило, действуют в других государствах Сообщества по принципу взаимного признания.
Важной особенностью Европейской системы является представление заключений независимых экспертов по основным разделам досье: «Качество», «Безопасность» и «Эффективность» вместе с заявкой на регистрацию. Такой порядок позволяет самым существенным образом сократить объем работы регистрационных органов в части экспертизы материалов заявок.
Другими особенностями европейской модели являются:
- подраздел «Фармацевтическая разработка» раздела «Качество», в котором излагаются основания выбора прописи дозированной формы,
- оценка спецификаций в разделе досье: «Качество» с учетом требований Европейской фармакопеи,
- специфические формы обеспечения качества субстанций, используемых в производстве дозированных форм: - Европейский вариант «мастер-файла» (DMF) и «сертификаты Европейской фармакопеи».
В США, строго говоря, системы регистрации нет, поскольку отсутствует единый регистр или реестр разрешенных препаратов. По этой причине неизвестно, сколько всего наименований было допущено на американский рынок. Действующая в стране система допуска новых лекарств в обращение теоретически распространяется лишь на рецептурные препараты, предназначенные для торговли между отдельными штатами федерального государства. На практике же эти отличия можно считать несущественными. Процедуру допуска фактически проходят нерецептурные препараты. По понятным причинам любой спонсор нового препарата стремится выйти на весь американский рынок, не ограничиваясь пределами какого-либо одного штата, а потому направляет заявку в соответствующий федеральный орган.
Американскими требованиями к материалам заявки не предусмотрен подраздел «Фармацевтическая разработка» раздела «Качество». Не требуется заключений независимых от спонсора экспертов по основным разделам досье. Общая оценка (экспертиза) заявок осуществляется чиновниками, а не экспертными органами, как во всех других странах.