
Молекулярно-генетическая диагностика и дифференцированная терапия гистиоцитарных пролиферативных заболеваний у детей 14. 01. 08 педиатрия 14. 01. 21 гематология и переливание крови
| Вид материала | Автореферат |
- Молекулярно-генетическая природа первичных гемофагоцитарных лимфогистиоцитозов в россии, 381.26kb.
- Аллогенная трансплантация гемопоэтических стволовых клеток в лечении врожденных и приобретенных, 1375.99kb.
- Факторы риска и контроль вирусных инфекций после трансплантации гемопоэтических стволовых, 767.07kb.
- Красняков Владимир Кириллович Совершенствование донорства крови и ее компонентов, 921.41kb.
- Прямое переливание крови (методические рекомендации), 154.15kb.
- Оптимизация инновационных технологий трансфузионного пособия пациентам регионального, 1245.79kb.
- Неопухолевые лимфаденопатии. 14. 00. 29 гематология и переливание крови, 1061.02kb.
- Обеспечение качества получения и клинического применения компонентов крови в субъекте, 404.49kb.
- кальцинированный аортальный стеноз-состояние системного гемостаза и реологических, 538.36kb.
- Иммуногематологическая оценка методов гемокомпонентной терапии у онкологических больных, 340.82kb.
На правах рукописи
Масчан Михаил Александрович
Молекулярно-генетическая диагностика и дифференцированная терапия гистиоцитарных пролиферативных заболеваний у детей
14.01.08 педиатрия
14.01.21 гематология и переливание крови
АВТОРЕФЕРАТ
Диссертации на соискание ученой степени
доктора медицинских наук
Москва 2011
Работа выполнена в ФГУ «Федеральный научно-клинический центр детской гематологии, онкологии и иммунологии» министерства здравоохранения и социального развития российской федерации
Научные консультанты:
Член-корреспондент РАМН, доктор медицинских наук, профессор
Румянцев Александр Григорьевич
Доктор медицинских наук
Новичкова Галина Анатольевна
Официальные оппоненты:
Доктор медицинских наук, профессор Алексеева Екатерина Иосифовна
Доктор медицинских наук, профессор Сметанина Наталья Сергеевна
Доктор медицинских наук, профессор Лукина Елена Алексеевна
Ведущая организация:
Санкт-петербургский государственный медицинский университет им. академика И.П.Павлова
Защита диссертации состоится « » 2011 года в часов на заседании диссертационного совета Д 208.050.01 в ФГУ ФНКЦ ДГОИ по адресу – 117997, Москва, ул. Саморы Машела, д.1
С диссертацией можно ознакомиться в библиотеке ФГУ ФНКЦ ДГОИ
Автореферат разослан « » 2011 года
Ученый секретарь диссертационного совета
доктор медицинских наук, профессор В.М.Чернов
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность проблемы Гистиоцитарные пролиферативные расстройства, или гистиоцитозы, - группа редких заболеваний, морфологической основой которых является патологическая пролиферация клеток гистиоцитарного ряда: тканевых макрофагов, моноцитов, дендритных клеток и их предшественников. Согласно современной классификации гистиоцитарных заболеваний, выделяют гистиоцитозы с вариабельным клиническим течением, основными представителями которых являются гистиоцитоз из клеток Лангерганса (ГКЛ) и гемофагоцитарный лимфогистиоцитоз (ГЛГ), и злокачественные гистиоцитозы, к которым относят моноцитарные варианты острого миелобластного лейкоза, солидные опухоли из моноцитов и дендритных клеток и хронический миеломоноцитарный лейкоз (ХММЛ) (современный термин - ювенильный миеломоноцитарный лейкоз (ЮММЛ)) (B.Favara, 1998). Несмотря на различия в биологии этих заболеваний, рассмотрение гистиоцитозов как единой клинико-патологической группы обусловлено сходной клинической презентацией у детей раннего возраста, необходимостью дифференциальной диагностики и аналогией методов терапии. Настоящее исследование сосредоточено на принципиальных вопросах биологии, диагностики и терапии трех наиболее распространенных форм гистиоцитарных расстройств у детей: первичного гемофагоцитарного лимфогистиоцитоза (ПГЛГ), ЮММЛ и ГКЛ.
ПГЛГ – врожденное расстройство иммунорегуляции, обусловленное генетическим дефектом механизмов клеточной цитотоксичности (G.Janka 2007, J-I.Henter 2002). Идентифицировано четыре гена, PRF1, UNC13D, STX и STXBP, герминальные мутации которых ведут к формированию клинического фенотипа ПГЛГ. Сходный клинический фенотип формируется у пациентов с Х-сцепленным лимфопролиферативным синдромом (Х-ЛПС) I и II типа и синдромом Грисселли, в основе которых лежат мутации в генах SH2D1A, XIAP и RAB27 соответственно (J.Pachlopnik Shmid, 2010). В отсутствие терапии ПГЛГ является смертельным заболеванием. Современная терапия ПГЛГ, основанная на последовательном применении иммуносупрессивной терапии (ИСТ) и аллогенной трансплантации гемопоэтических стволовых клеток (ТГСК), позволяет излечивать около 50% пациентов (J-I.Henter, 2007). Своевременная верификация генетической природы ПГЛГ позволяет подтвердить показания к ТГСК, выполнить генетическое консультирование и пренатальную диагностику. Исследование генетической эпидемиологии ПГЛГ в популяции российских пациентов, анализ эффективности терапии ПГЛГ и разработка алгоритма клинической и лабораторной диагностики необходимы для улучшения диагностики и результатов лечения этого заболевания
ЮММЛ – заболевание, сочетающее черты миелодисплазии и миелопролиферации (M.Arico, 1997). В основе заболевания лежат соматические (реже - герминальные) мутации в генах PTPN11, NRAS, KRAS, CBL, NF1 (M.Loh, 2010). Результатом является гиперпролиферация миелоидных и моноцитарных предшественников, дисфункция костномозгового кроветворения и патологическая инфильтрация органов. Единственным методом, позволяющим излечивать пациентов с ЮММЛ, считается ТГСК. Выполнение ТГСК при ЮММЛ ассоциировано с 10-20% риском трансплантационной смертности и 30-40% риском рецидива заболевания (F.Locatelli, 2005). Нерешенным остается вопрос о месте нетрансплантационных методов терапии, таких как дифференцировочная терапия (ДТ) 13-цис-ретиноевой кислотой (13-RA) и высокодозная химиотерапия (ВХТ) (M.Loh, 2010). Разработка алгоритма молекулярно-генетического анализа и клинико-генетическое сопоставление необходимы для диагностики, определения показаний к ТГСК, мониторинга минимальной остаточной болезни и тестирования новых методик терапии. Анализ результатов ТГСК и альтернативной терапии необходим для оптимизации технологии ТГСК и определения перспективной тактики мультимодальной терапии.
ГКЛ – заболевание, в основе которого лежит клональная пролиферация миелоидных дендритных клеток, фенотипически сходных с эпидермальными клетками Лангерганса. Манифестация ГКЛ варьирует от локализованных очагов остеолизиса до диссеминированных форм с неконтролируемым злокачественным течением. Биологическая основа этих различий и природа ГКЛ в целом остаются нерасшифрованными (M.Egeler, 2010). Данные о роли мутации V600E в гене BRAF в патогенезе ГКЛ стали новым фактом, свидетельствующим в пользу неопластической природы заболевания (G.Badalyan-Very, 2010). Подтверждение этого факта может открыть новый этап понимания биологии заболевания и новые перспективы терапии. Современным стандартом лечения ГКЛ у детей является комбинированная терапия винбластином (Vbl) и преднизолоном (Prn) (M.Minkov, 2011). Одной из наиболее актуальных проблем является терапия пациентов группы «высокого риска» - пациентов с мультисистемным ГКЛ с вовлечением «органов риска» (МСОР+). Общая выживаемость в этой группе не превышает 70%, а в подгруппе пациентов, рефрактерных к стандартной терапии – 20% (M.Minkov, 2002, H.Gadner, 2001). В лечении резистентных форм ГКЛ наиболее перспективным представляется опыт интенсивной химиотерапии (ХТ) с использованием 2-хлор-дезоксиаденозина (2-CdA) в комбинации с цитозина арабинозидом (AraC) (F.Bernard, 2005). Таким образом, исследование молекулярного дефекта V600E BRAF при ГКЛ, анализ результатов терапии мультисистемного ГКЛ и разработка альтернативных программ терапии являются актуальным прикладным вопросом детской гематологии/онкологии и педиатрии.
Цель исследования
Разработка и внедрение современных принципов клинико-лабораторной и молекулярно-генетической диагностики и оптимизирующих программ терапии пациентов с первичным гемофагоцитарным лимфогистиоцитозом, ювенильным миеломоноцитарным лейкозом и гистиоцитозом из клеток Лангерганса.
Задачи исследования
- Выполнить молекулярно-генетический анализ генов PRF1, UNC13D, STX, STXBP, SH2D1A, XIAP, RAB27 в группе российских пациентов с клиническим диагнозом ПГЛГ. Изучить клиническую презентацию и корреляцию генотип-фенотип при генетически детерминированном ГЛГ.
- Разработать алгоритм клинико-лабораторной диагностики, молекулярно-генетической верификации и дифференциальной диагностики первичного гемофагоцитарного синдрома и его генетических субвариантов.
- Провести анализ результатов лечения пациентов с ПГЛГ с использованием стандартных режимов программной иммуносупрессивной химиотерапии и алло-ТГСК. Разработать практические рекомендации по выбору тактики терапии.
- Выполнить молекулярно-биологический анализ генов PTPN11, NRAS, KRAS и CBL в репрезентативной группе пациентов с ЮММЛ. Сопоставить клинические и лабораторные проявления с генетическими дефектами и разработать рациональную тактику генетической верификации диагноза и алгоритм клинико-лабораторной диагностики и дифференциальной диагностики ЮММЛ.
- Провести анализ результатов алло-ТГСК, ВХТ и ДТ с использованием 13-RA и в группе пациентов с ЮММЛ. Разработать клиническую стратификацию на основании анализа клинических и лабораторных факторов риска. Разработать стратегию выбора терапии ЮММЛ в соответствии с клинико-биологичекой стратификацией.
- Исследовать мутацию V600E в гене BRAF в патогистологических образцах пациентов с гистиоцитозом из клеток Лангерганса. Оценить потенциальное место исследования мутационного статуса BRAF в диагностике, выборе и мониторинге терапии ГКЛ.
- Изучить непосредственные и отдаленные результаты стандартной терапии пациентов с ГКЛ. Разработать альтернативный подход к лечению пациентов с мультисистемным заболеванием на основе использования комбинированной ХТ 2-CdA и AraC. Провести сравнительный анализ результатов стандартной и альтернативной терапии. Разработать практические рекомендации по выбору терапии и тактике сопроводительной терапии ГКЛ в группе высокого риска.
Научная новизна
Впервые в репрезентативной выборке российских пациентов с ПГЛГ выполнено молекулярно-генетическое исследование всех известных генов ( PRF1, UNC13D, STX, STXBP, SH2D1A, XIAP, RAB27), ассоциированных с фенотипом ПГЛГ. Выявлено уникальное распределение генетических вариантов ПГЛГ с преобладанием FHL3, отличное от популяций с другим этническим составом. Показано, что распространение в популяции мутаций c.2346_2349del и c.3037insG обусловлено «эффектом основателя». Не выявлена значимая корреляция клинического фенотипа с генетическим вариантом ПГЛГ. В работе проведен первый в России анализ результатов терапии пациентов с ПГЛГ и убедительно продемонстрирована необходимость выполнения ТГСК всем пациентам с ПГЛГ. Показано, что основным препятствием к успеху ТГСК является высокая трансплантационная смертность (54 ± 15%). Впервые в репрезентативной выборке российских пациентов с ЮММЛ выполнено молекулярно-генетическое исследование основных генов (PTPN11, NRAS, KRAS и CBL), ассоциированных с развитием ЮММЛ. В работе подтверждено характерное распределение генетических вариантов ЮММЛ и впервые выявлена корреляция клинического фенотипа с генотипом заболевания. Разработана программа лечения 13-RA и низкими дозами AraC при ЮММЛ. Продемонстрирован клинико-гематологический эффект ДТ и установлена корреляция исходных клинико-лабораторных показателей и генетического варианта ЮММЛ с ответом на ДТ. Подробно описан феномен стойкого ответа на ДТ, выявлена персистенция мутантного клона у пациентов в статусе полного ответа (ПО) и описано развитие поздних злокачественных (острый лимфобластный лейкоз (ОЛЛ) и аутоиммунных (тромботическая тромбоцитопеническая пурпура (ТТП)) заболеваний у пациентов с ПО на ДТ. Впервые в репрезентативной группе российских пациентов проведен анализ результатов ТГСК, 5-летняя общая выживаемость составила 3813%, трансплантационная смертность - 2812%, частота развития рецидива 31%. Выполнен анализ мутации V600E BRAF у пациентов с ГКЛ и подтверждена вероятная роль данного соматического генетического дефекта в патогенезе ГКЛ у 24% пациентов. Разработана оригинальная программа терапии для группы пациентов «высокого риска» на основе использования комбинированной терапии 2-CdA и AraC. Показана высокая эффективность терапии рефрактерных форм МСОР+ ГКЛ, рОВ = 6615%. Показана высокая эффективность 2-CdA и AraC в лечении МСОР+ ГКЛ, рОВ = 889%, частота развития инвалидизирующих перманентных последствий – 0%. Описан необычный спектр инфекционных осложнений, включающий инфекции вакцинальным штаммом микобактерий M.bovis и тяжелые инфекции, вызванные респираторными вирусами.
Практическое значение
В работе на основании анализа генетической структуры ПГЛГ в группе российских пациентов разработан алгоритм клинико-лабораторной диагностики ПГЛГ, алгоритм молекулярно-гентического анализа и методика выявления наиболее распространенных мутаций в гене UNC13D. На основании анализа результатов ИСТ и ТГСК предложена стратегия выбора терапии, предусматривающая начало поиска донора в момент установления диагноза ПГЛГ и обязательное выполнение ТГСК не позднее 4 месяцев от момента установления диагноза. Разработан алгоритм молекулярно-генетического исследования при диагностике пациентов с клиническим диагнозом ЮММЛ, позволяющий верифицировать диагноз, осуществить рациональный выбор терапии и мониторинг заболевания после ТГСК. Разработан алгоритм выбора терапии в соответствии с клинико-биологической стратификацией пациентов. На основании анализа результатов ХТ и ТГСК предложена стратегия выбора терапии, предусматривающая начало поиска донора в момент установления диагноза ЮММЛ и обязательное выполнение ТГСК не позднее 6 месяцев от момента установления диагноза в группе пациентов, не ответивших на ДТ. Молекулярно-генетический анализ мутации V600E BRAF у пациентов с ГКЛ показал, что данная генетическая аномалия потенциально является диагностическим маркером заболевания и терапевтической мишенью. Разработана и внедрена в клиническую практику эффективная программа ВХТ на основе комбинации 2-CdA и AraC для лечения пациентов с рефрактерным течением ГКЛ МСОР+. Разработаны рекомендации по сопроводительной терапии.
Основные положения, выносимые на защиту
- Гистиоцитарные пролиферативные расстройства – гистиоцитозы – группа фенотипически родственных заболеваний, в основе которых лежит спектр врожденных либо приобретенных молекулярных дефектов, ведущих к аномальной пролиферации и регуляции функциональной активности клеток гистиоцитарного ряда. Новые данные о биологии гистиоцитозов диктуют необходимость разработки комплексного алгоритма диагностики, дифференцированной тактики терапии и диспансерного наблюдения пациентов с гистиоцитозами.
- Мутации в гене UNC13D являются наиболее распространенным генетическим дефектом в популяции российских пациентов с ПГЛГ. Частота выявления мутаций в гене UNC13D составила 54%. Распространенность этих мутаций в исследованной группе обусловлена «эффектом основателя» - персистенцией в популяции мутаций c.2346_2349del и c.3037insG. Исследование гена UNC13D рекомендуется в качестве первого этапа молекулярно-генетической диагностики ПГЛГ в России.
- Единственным методом терапии, обеспечивающим долгосрочную выживаемость пациентов с ПГЛГ, является ТГСК. 5-летняя рОВ в группе ПГЛГ составляет 37±14 % при выполнении ТГСК, 0±0% при выполнении только ИСТ, р = 0,0005. Неудачи ТГСК обусловлены высокой трансплантационной смертностью, рTRM = 54±15%, Идентификация и выбор донора являются приоритетными задачами наравне с генетической верификацией диагноза и началом иммуносупрессивной терапии. ТГСК должна быть выполнена не позднее 4 месяцев от начала ИСТ, так как частота реактивации заболевания составляет 95%, медиана реактивации – 5 месяцев.
- Молекулярно-генетическое исследование генов PTPN11, NRAS, KRAS и CBL позволяет верифицировать диагноз у 70% пациентов с ЮММЛ. Относительная частота различных генетических дефектов составляет: PTPN11 - 35%, NRAS - 14%, CBL - 13%, KRAS - 11%. Стратификация пациентов на основании генетического варианта ЮММЛ (мутации в генах RAS и CBL), возраста манифестации (< 1 года) и содержания фетального гемоглобина (HbF) (< 10%) позволяет выделить прогностически благоприятную группу, не нуждающуюся в выполнении ТГСК в первой линии терапии. Пациентам этой группы показано проведение ДТ 13-RA + AraC, 5-летняя рОВ при проведении ДТ составляет 75±15%.
- Пациенты группы высокого риска и пациенты, не ответившие на терапию 13-RA + AraC, нуждаются в выполнении ТГСК по витальным показаниям не позднее 6 месяцев от установления диагноза. ТГСК должна быть выполнена независимо от наличия полностью совместимого донора. 5-летняя рОВ при выполнении ТГСК составила 38±13%. Применение флударабина (Flu) в комбинации с бусульфаном (Bu) и мельфаланом (Mel) обеспечивает эквивалентную частоту приживления трансплантата (90% и 60%, р = 0.24) и общую выживаемость (рОВ 47±19% и 33±19%, р = 0.72) в сравнении с стандартным режимом кондиционирования: циклофосфамид (Cy) + Bu + Mel.
- Мутация V600E BRAF выявляется у 24% пациентов с ГКЛ и, вероятно, принимает участие в патогенезе заболевания. Значимой корреляции статуса BRAF с клиническими и лабораторными проявлениями заболевания не выявлено. Мутация V600E BRAF потенциально является диагностическим маркером и терапевтической мишенью у пациентов с ГКЛ.
- Комбинированная ХТ 2-Сda и AraC является эффективным методом лечения мультисистемного ГКЛ, резистентного к стандартной терапии, рОВ = 66±19%. Применение 2-Сda и AraC в терапии первой линии МСОР+ ГКЛ более эффективно в сравнении со стандартной терапией. Частота достижения полных ответов составила 100% vs. 33%, рБСВ = 88±9% vs 40±11%, частота формирования перманентных осложнений, 66% vs 0%. Достоверных различий рОВ не выявлено, что обусловлено высокой эффективностью терапии второй линии. Применение ВХТ 2-Сda + AraC ассоциировано с выраженной миелосупрессией и иммуносупрессией и высоким риском развития инфекционных осложнений. Характер иммуносупрессии определяет спектр инфекций, включающий вирусные инфекции и инфекции вакцинальным штаммом M.bovis.
Внедрение в практику
Результаты работы внедрены в практику молекулярной диагностики ПГЛГ в Медико-генетическом научном центре РАМН (зав. лабораторией профессор А.В.Поляков) и диагностики ЮММЛ в лаборатории молекулярной биологии ФНКЦ ДГОИ (зав. лабораторией к.м.н. Бобрынина В.О.). Алгоритмы диагностики и оптимизированные протоколы терапии используются в отделениях гематологии и трансплантации костного мозга РДКБ и в клинике ФНКЦ ДГОИ. Основные положения и выводы диссертации используются в курсе лекций и семинаров по детской гематологии/онкологии ФУВ РГМУ им. Н.И. Пирогова
Апробация диссертации
Материалы диссертации представлены в докладах на национальных и международных конференциях, в частности на конференции международного общества по изучению гистиоцитозов (Histiocyte Society) в 2005, 2008, 2009 гг., международного общества детских онкологов (SIOP) в 2010 г., европейской ассоциации гематологии (EHA) в 2010 году, европейского общества генетики человека (ESHG) в 2008, 2009, 2010 гг., российском национальном конгрессе «Человек и лекарство» в 2011 году. Основные положения и выводы диссертации используются в курсе лекций и семинаров по детской гематологии/онкологии ФУВ РГМУ им. Н.И.Пирогова. Материалы диссертации опубликованы в составе 25 печатных работ, в том числе 10 в изданиях, рекоммендованых ВАК.
Апробация диссертации состоялась 15 марта 2011 г. на научно-практической конференции ФНКЦ ДГОИ.
Структура и объем диссертации
Материал диссертации изложен в 8 главах. Структура диссертации традиционна и включает разделы: введение, обзор литературы, пациенты и методы, результаты, обсуждение, выводы и практические рекоммендации. Объем диссертации составляет 400 страниц. Диссертация включает 40 рисунков и 35 таблиц.
Содержание работы
Пациенты и методы
Формирование выборки. В исследование включено 180 пациентов в возрасте от 1 месяца до 17 лет с диагнозом ПГЛГ, ЮММЛ и ГКЛ. В группу ПГЛГ включены 34 пациента из 31 семьи. Молекулярно-генетическое исследование выполнено 26 пациентам. Анализ результатов терапии выполнен в группе 32 пациентов, 2 пациента с верифицированным диагнозом Х-ЛПС были исключены. В группу ЮММЛ включен 61 пациент, молекулярно-генетическое исследование выполнено 44 пациентам, анализ результатов терапии выполнен во всей группе. В группу ГКЛ включены 86 пациентов, в анализ результатов терапии включены 48 пациентов, молекулярно-генетическое исследование выполнено 37 пациентам. Основу выборки составили все пациенты (n = 143) с соответствующим диагнозом, наблюдавшиеся в отделении общей гематологии РДКБ в период с 1993 по 2010 гг. Молекулярно-генетическое исследование ГКЛ выполнено на биоматериале пациентов из различных клиник, архивные образцы которых были доступны для анализа.
Критерии диагноза. Диагноз ПГЛГ и ЮММЛ устанавливали на основании международных критериев приведенных в табл. 1 и 2. Диагноз ГКЛ устанавливали в соответствии с рекомендациями The Histiocyte Society на основании гистологической картины и иммуногистохимической детекции маркера CD1a на патологических клеточных элементах. Клиническое стадирование пациентов с ГКЛ проводили в соответствии с рекомендациями The Histiocyte Society. В соответствии с числом пораженных органов и систем выделяли моносистемное (МоноС) заболевание и мультисистемное (МС) заболевание. При мультисистемном заболевании выделяли формы с вовлечением «органов риска» (МСОР+) и без такового (МСОР-). К «органам риска» относили печень, кроветворную систему, селезенку и легкие.
Обследование пациентов. Минимальный объем клинико-лабораторного обследования включал: 1) автоматический анализ крови с подсчетом лейкоцитарной формулы; 2) биохимический анализ крови с определением АЛТ, АСТ, ЩФ, ЛДГ, билирубина, мочевины, креатинина, альбумина; 3) коагулограмму; 4) миелограмму с окраской гематоксилин-эозин; 5) иммуноглобулины сыворотки; 6) УЗИ брюшной полости; 7) рентгенографию грудной клетки; 8) общий анализ мочи. Дополнительный набор исследований для пациентов с ПГЛГ включал: 1) триглицериды сыворотки (натощак); 2) ферритин сыворотки; 3) исследование спинномозговой жидкости с определением цитоза, содержания белка, лейкоцитарной формулы цитопрепарата. Для пациентов
| Таблица 1 Диагностические критерии гемофагоцитарного лимфогистиоцитоза, Histiocyte Society, 2004 | |
| ³ 38,50 > 7 дней |
| > 3 см из под края рёберной дуги |
| в ≥ 2-х линиях |
| |
|
|
|
| ³500 мкг/л |
| ³ 2500 Ед/л |
| |
| |
| Для установления диагноза необходимо выполнение 5 из 8 критериев либо молекулярно-генетическая верификация диагноза. | |
с ЮММЛ: 1) электрофорез гемоглобина; 2) цитогенетическое/молекулярно-генетическое исследование костного мозга с целью выявления Ph-хромосомы/химерного транскрипта BCR-ABL; 3) культуральное исследование периферической крови с оценкой спонтанного колониеобразования гранулоцитарно-макрофагальных колоний. Для пациентов с ГКЛ включал: 1) пробу Зимницкого; 2) обзорную рентгенографию скелета или радиоизотопное исследование с Технецием-99; 3) биопсию патологического очага.
Молекулярно-генетическое исследование. В группе пациентов с ПГЛГ исследованы все экзоны генов, ассоциированных с клиническим фенотипом ПГЛГ: PRF1, UNC13D, STX, STXBP, SH2D1A и XIAP. В группе ЮММЛ для исследованы экзоны 3 и 13 гена PTPN11, 2 и 3 генов NRAS и KRAS, 8 и 9 гена CBL. Молекулярно-генетическое исследование ГКЛ было ограничено мутацией V600E BRAF. Прямое автоматическое секвенирование в группе ПГЛГ: геномную ДНК выделяли с помощью коммерческого набора DNA Prep 100 Diatom TM из образцов периферической крови. Амплификацию фрагментов ДНК проводили методом ПЦР на термоциклере МС2 фирмы «ДНК-технология» (Россия). Прямое автоматическое секвенирование в группе ЮММЛ: геномную ДНК выделяли с помощью набора «ДНК-сорб-В», АмплиСенс, ФГУН «ЦНИИЭ» Роспотребнадзора, Москва, Россия, из образцов периферической крови. Амплификацию фрагментов ДНК проводили методом ПЦР на программируемом термоциклере Dyad, BioRad. Все фрагменты секвенированы с обеих цепей ДНК с использованием Big Dye Terminator’s v 1.1 Cycle Sequencing Kit и анализатора ABI PRISM 3130xl (Applied Biosystems, Foster City, CA, США), анализ результатов проводили с
| Таблица 2 Критерии диагностики ювенильного миеломоноцитарного лейкоза [M.Loh, 2010] | |
|
|
| |
| |
| |
|
|
| |
| |
|
|
| |
| |
| |
| |
| Диагностическое правило: необходимо выполнение всех критериев группы I и одного из критериев группы II. При отсутствии критериев группы II, необходимо выполнение двух критериев группы III. | |
помощью программы ChromasPro. При исследовании гаплотипа основателя для повторяющихся мутаций в гене UNC13D были выбраны 8 микросателлитных маркеров на хромосоме 17q25, в области размером 7,4 млн.п.н. вокруг исследуемого гена. Для каждого из маркеров были выбраны и пары праймеров. Результаты оценивали с помощью электрофореза в полиакриламидном геле с окрашиванием раствором бромистого этидия и регистрацией с помощью системы GelDoc, BioRad, США. Выделение клеточных линий из нуклеарной фракции периферической крови проводили реагентами для иммуномагнитной селекции производства Dynal, Invitrogen, США, в соответствии с инструкцией производителя. Исследование мутации V600E BRAF при ГКЛ: ДНК выделялась из биоптатов, фиксированных и залитых в парафин, после макродиссекции. После депарафинизации ксилолом и проводки по спиртам проводилась инкубация с протеиназой К в течение ночи и фенол-хлороформная экстракция. Для выявления мутации проводилась ПЦР в реальном времени с использованием флюоресцентных зондов TaqMan. Дизайн праймеров проводился таким образом, чтобы избежать неспецифической амплификации псевдогена на Х хромосоме. Зонд, специфичный к нормальной последовательности гена был помечен флуорофором FAM на 5’ конце, специфичный мутантной последовательности – HEX. Для повышения чувствительности метода в нуклеотидный состав обоих зондов были введены LNA (locked nucleic acids). Амплификация проводилась на приборе CFX-96, BioRad. Данные флюоресценции анализировались с помощью программы аллельной дискриминации. Все образцы амплифицировались отдельно с использованием другой пары праймеров, после чего выполнялось прямое секвенирование на приборе AB3500, Applied Biosystems.
Определение статуса заболевания, критерии ответа на терапию и сроки оценки ответа
Первичный гемофагоцитарный лимфогистиоцитоз Частичный ответ (ЧО) – отсутствие лихорадки >37,7oC, сокращение размеров селезенки, тромбоциты >100х109/л, фибриноген > 1,5 г/л. Полный ответ (ПО) – отсутствие лихорадки >37,7oC, нормальный размер селезенки, нормализация показателей периферической крови (гемоглобин >100г/л, тромбоциты >100х109/л, нейтрофилы >0,5х109/л), триглицериды < 2 ммоль/л, ферритин < 500мкг/л, нормальные показатели СМЖ. Активное заболевание (АЗ) – пациенты, не выполняющие критерии ПО и ЧО. Реактивация заболевания (РЗ) - появление ≥3 признаков из 7 (лихорадка >38,5oC, тромбоциты <100х109/л, спленомегалия, триглицериды >2 ммоль/л, фибриноген <1,5 г/л, гемофагоцитоз, ферритин > 500мкг/л) после достижения ПО или ЧО. Появление неврологического дефицита или характерных изменений состава спинномозговой жидкости (СМЖ) достаточно для констатации реактивации заболевания. Ответ на ИСТ оценивали через 2 месяца от начала лечения.
Ювенильный миеломоноцитарный лейкоз Общепринятых критериев оценки ответа на терапию при ЮММЛ не существует. В настоящей работе были приняты следующие определения статуса заболевания: полный ответ (ПО) - разрешение всех клинических и лабораторных проявлений заболевания, не поддерживаемое терапией в течение > 1 месяца.Частичный ответ (ЧО) – лейкоциты < 10x109/л, тромбоциты > 100x109/л, гемоглобин > 100 г/л, сокращение размеров печени (≤ 3 см из-под ребра) и селезенки (≤ 3 см из-под ребра), отсутствие специфической инфильтрации другой локализации. Отсутствие бластемии. Стабилизация заболевания (СЗ) – снижение лейкоцитоза на ≥ 50% от исходного, повышение уровня тромбоцитов на ≥ 50% от исходного, сокращение размеров печени и селезенки на ≥ 50% от исходного. Прогрессия заболевания (ПЗ) - сохранение лабораторных и клинических проявлений заболевания при невыполнении условий СЗ. Совокупно ПО и ЧО рассматривался как клинически значимый ответ, СЗ и ПЗ как отсутствие ответа. Ответ на терапию оценивали не ранее, чем через 2 месяца от начала терапии.
Гистиоцитоз из клеток Лангерганса Полный ответ (ПО) – разрешение всех обратимых клинических и лабораторных проявлений заболевания. Частичный ответ (ЧО) – неполное разрешение исходных клинических и лабораторных проявлений заболевания. Прогрессия заболевания (ПЗ) - появление новых очагов поражения и/или сохранение и/или ухудшение со стороны исходных очагов/клинико-лабораторных проявлений. Перманентные осложнения – необратимые изменения, развившиеся вследствие поражения органа/ткани ГКЛ: несахарный диабет, фиброз легких, фиброз/цирроз печени, гипопитуитаризм. Реактивация заболевания (РЗ) – появление клинических и лабораторных признаков заболевания после констатации достижения ПО. Сроки оценки ответа – ответ на терапию оценивался по завершении интенсивной фазы терапии (6 недель и 12 недель при терапии по стандартным протоколам) или после каждого блока ВХТ (пилотный протокол).
Терапия
Первичный гемофагоцитарный лимфогистиоцитоз
Терапию ПГЛГ проводили в соответствии с рекомендациями протокола HLH-94 (n=24) и HLH-2004 (n=5), 3 пациента получили сопроводительную терапию. Терапия состояла из ИСТ и ТГСК.
Иммуносупрессивная терапия В состав ИСТ был включен этопозид (VP-16), дексаметазон (Dexa), циклоспорин А (CsA), метотрексат (MTX)(эндолюмбально) и Prn (эндолюмбально). Рис. 1. При развитии реактивации заболевания назначали ИСТ второй линии по выбору врача. Антитимоцитарный глобулин (АТГ), АТГАМ, Пфайзер, США в дозе 160 мг/кг/курс, получили 2 пациента. Комбинированную ВХТ в составе Тиофосфамид (TT) 2 мг/кг, винкристин (Vcr) 1,5 мг/м2, флударабин (Flu) 150 мг/м2, Prn 5 мг/кг/сутки получили 4 пациента. Циклофосфамид (Cy) в дозе 500 мг/м2 получили 2 пациента. Повторную терапию элементами стандартного протокола получили 12 пациентов.*
Трансплантация гемопоэтических стволовых клеток ТГСК выполнена 12 пациентам. Восьми пациентам проводили миелоаблативное кондиционирование, 4 пациентам – кондиционирование со сниженной интенсивностью. У 3 пациентов ТГСК выполнена от HLA-совместимого сиблинга (MSD), у 7 пациентов – от HLA-совместимого неродственного донора (MUD), у 2 пациентов – от частично совместимого родственного донора (MMRD). Профилактику реакции «трансплантат против хозяина» (РТПХ) проводили ингибитором кальциневрина (CsA, n = 7 или такролимус (Tacro), n = 4) в комбинации с MTX (n = 3) или мофетила микофенолятом (MMF) (n = 7). Один пациент получал MTX и MMF. У 1 пациента для профилактики РТПХ выполнили селекцию CD34+-клеток на приборе CliniMacs, Miltenyi Biotec, Германия. Детали процедуры трансплантации представлены в табл.3.
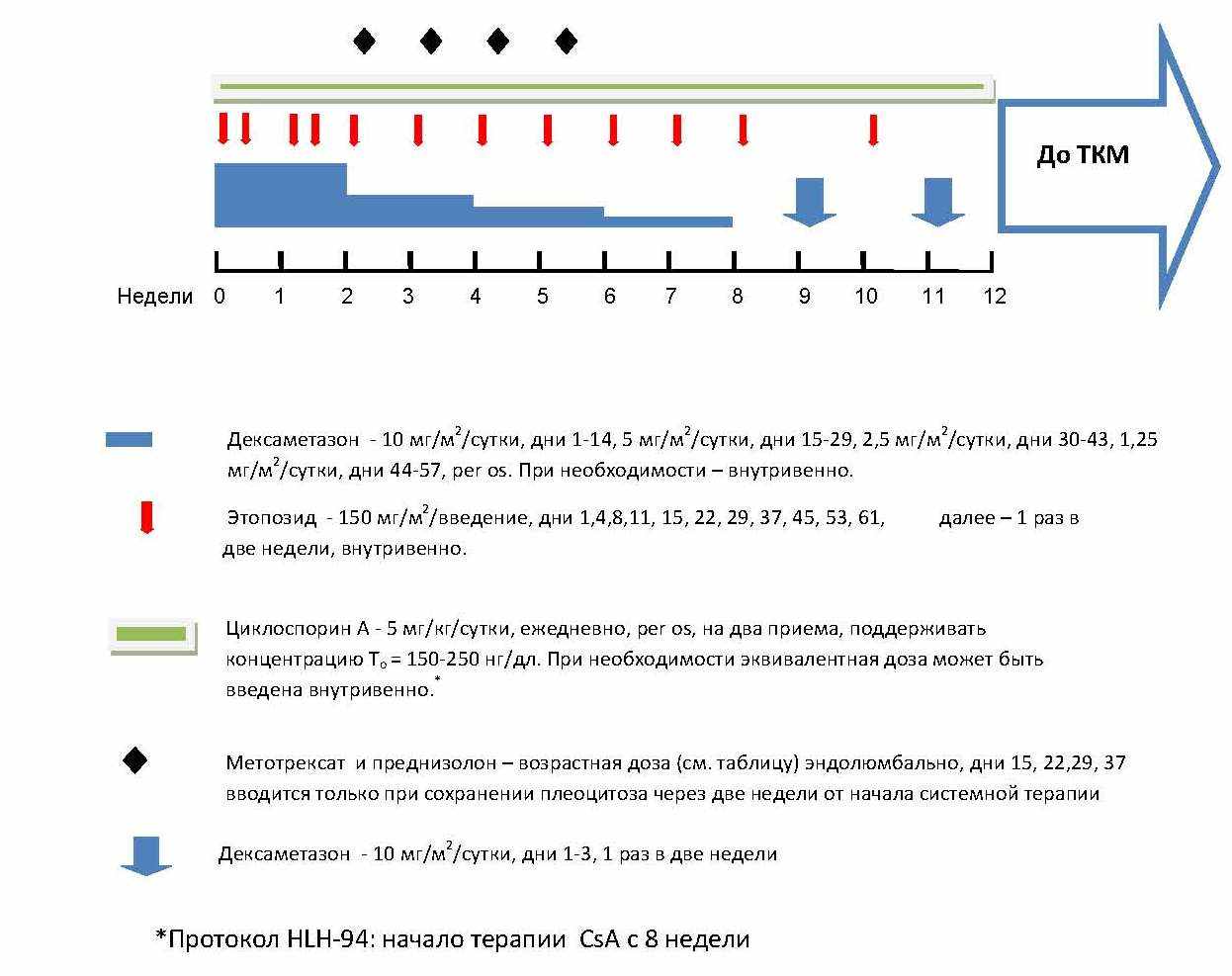 |
| Рисунок 1 Схема терапии ПГЛГ в соответствии с протоколом HLH-2004 |
Ювенильный миеломоноцитарный лейкоз
В первой линии терапии пациенты с ЮММЛ получали ДТ (n = 32) или ВХТ (n = 18), ТГСК (n = 1), сопроводительную терапию (n = 10).
Дифференцировочная терапия. ДТ включала 13-RA в дозе 100 мг/м2/сутки ежедневно в комбинации с AraC в дозе 10-30 мг/м2/сутки 10-14 дней каждого месяца. Дозу AraC корректировали для поддержания концентрации лейкоцитов в интервале 3-10х109/л.
Высокодозная химиотерапия. ВХТ проводили в соответствии со стандартными элементами программной терапии острых миелобластных лейкозов. Блоки FLA (Flu в дозе 30 мг/м2 №5, AraC в дозе 2000 мг/м2 №5), FLAM (FLA + митоксантрон (Mit) в дозе 10-12 мг/м2 №3), FLAIda (FLA + идарубицин (Ida) в дозе 8-10 мг/м2 №3), ADE ( AraC 200 мг/м2/сутки №7, даунорубицин (DNR) 45-60 мг/м2 №3, VP-16 150 мг/м2 №3), AraCVp (AraC 500 мг/м2/сутки №5, VP-16 100 мг/м2 №5), IFOVP-16 (ифосфамид (IFO) 2000 мг/м2/сутки №5, VP-16 100 мг/м2 №5), AME (AraC 200 мг/м2/сутки №7, Mit 10-12 мг/м2 №3, VP-16 100-150 мг/м2 №3), HAE (AraC 2000-6000 мг/м2/сутки №4, VP-16 100 мг/м2 №4). При достижении ПО или ЧО на ДТ пациенты продолжали получать ДТ терапию до развития рецидива ЮММЛ, развития других гематологических заболеваний либо решения о выполнении ТГСК. При отсутствии ответа на ДТ пациенты получали ВХТ в качестве терапии второй линии. Трансплантация гемопоэтических стволовых клеток Аллогенная ТГСК выполнена 20 пациентам. Семнадцать пациентов получили ТГСК в отделении трансплантации костного мозга РДКБ и включены в детальный анализ результатов ТГСК, 3 пациента трансплантированы в других клиниках и включены только в анализ выживаемости. Все пациенты получили миелоаблативное кондиционирование. У 5 пациентов ТГСК выполнена от MSD, у 8 пациентов – от MUD, у 4 пациентов – от MMRD. В качестве источника ГСК использовали КМ (n = 7), СКПК (n = 7, из них в 3 случаях проводили селекцию CD34+-клеток на приборе CliniMacs, Miltenyi Biotec, Германия), ПК (n = 3). Профилактику РТПХ проводили ингибитором кальциневрина (CsA, n = 10 или Tacro, n = 6) в комбинации с MTX (n = 1) или MMF (n = 8) или Prn (n = 1). Один пациент получал MTX и MMF. Детали ТГСК суммированы в табл. 4
Гистиоцитоз из клеток Лангерганса
Стандартная терапия первой линии. В период с 1994 по 2003 год пациенты получали терапию первой линии в соответствии с рекомендациями одного из стандартных международных протоколов: DAL-HX-90 (n = 4), LCH I (n = 5), LCH II (n = 18), LCH III (n = 7). Дозы и порядок введения препаратов представлены на рис. 2.
Терапия второй линии. В случае неудачи терапии первой линии выбор терапии второй линии осуществлялся индивидуально, так как общепринятого стандарта терапии второй линии не существовало. Терапия второй линии суммирована в табл. 5. Пять пациентов получили терапию с использованием комбинации 2-CdA и промежуточных доз AraC.
-
Таблица 3 характеристики процедуры трансплантации в группе пациентов с первичным гемофагоцитарным лимфогистиоцитозом
Донор1
Совместимость
Источник2
NC, х108/кг
Кондиционирование3
Профилактика РТПХ4
18.GL3
UCB
8/10
ПК
0,8
Bu 19 мг/кг
Cy 120 мг/кг
Flu 100 мг/м2
Atgam 90 мг/кг
Tacro 0,03 мг/кг
MMF 30 мг/кг
29.GL32
UCB
8/10
ПК
3,4
Mel180 мг/м2
Flu 150 мг/м2
Thymo 10 мг/кг
Tacro 0,03 мг/кг
MMF 30 мг/кг
MTX4
31.GL2.1
MUD
10/10
СКПК
22
Bu 20 мг/кг
Cy 120 мг/кг
Flu 100 мг/м2
Atgam 90 мг/кг
CsA 3 мг/кг
MMF 30 мг/кг
4.GL
MUD
10/10
КМ
4,1
Bu 16 мг/кг
Cy 120 мг/кг
Flu 90 мг/м2
Atgam 90 мг/кг
MMF 30 мг/кг
MTX4
13.GL12
MUD
8/8
KM
8
Bu 20 мг/кг
Cy 200 мг/кг
Atgam 160 мг/кг
CsA 3 мг/кг
MTX 30 мг/кг
Prn 1 мг/кг
25.GL1
MUD
10/10
СКПК
12
Bu 20 мг/кг
Cy 120 мг/кг
Flu 100 мг/м2
Thymo 10 мг/кг
Tacro 0,03 мг/кг
MMF 30 мг/кг
28.GL10
MUD
10/10
СКПК
23
Bu 8 мг/кг
Cy 120 мг/кг
Flu 150 мг/м2
Atg-F 40 мг/кг
CsA 3 мг/кг
MMF 30 мг/кг
MTX4
2.GL27
MMRD
8/10
КМ
8,4
Mel 180 мг/м2
TT 10 мг/кг
Flu 150 мг/м2
Atgam 90 мг/кг
Tacro 0,03 мг/кг
MMF 30 мг/кг
17.GL11
MMRD
4/6
СКПК5
8
Bu 16 мг/кг
Cy 200 мг/кг
Flu 90 мг/м2
Atgam 90 мг/кг
CsA 3 мг/кг
21.GL9
MSD
6/6
СКПК
8,2
Bu 16 мг/кг
Cy 120 мг/кг
Flu 90 мг/м2
Atgam 90 мг/кг
CsA 3 мг/кг
MMF 30 мг/кг
14.GL8
MSD
6/6
СКПК
18
TT 15 мг/кг
Cy 15 мг/кг
Flu 90 мг/м2
Atgam 90 мг/кг
CsA 3 мг/кг
35GL
MSD
6/6
КМ
nd6
Bu 16 мг/кг
Cy200 мг/кг
Atgam 90 мг/кг
CsA 3 мг/кг
1 UCB - неродственная пуповинная кровь, MSD – совместимый сиблинг, MUD – неродственный совместимый донор, MMRD – родственный частично совместимый донор
2 ПК – пуповинная кровь, СКПК – стволовые клетки периферической крови, КМ – костный мозг
3 TT – тиофосфамид, Mel – мельфалан, Flu – флударабин, Bu – бусульфан, Thymo – тимоглобулин, Cy – циклофосфамид, Аtg-F – АТГ-Фрезениус,
ATGAM – атгам.
4 Tacro – такролимус, CsA – циклоспорин А, MMF – мофетила микофенолат, MTX– метотрексат, доза 15 мг/м2 день +3, 10 мг/м2 – день +6, +11, Prn –
преднизолон
5 СD34+ -селекция
6 nd- нет данных
| Таблица 5 Терапия второй линии в группе пациентов с ГКЛ, не ответивших на стандартную терапию первой линии | ||||||
| № | Возраст, месяцев | Терапия | Статус через 6 недель | Альтернативная терапия (до 2-CdA) | Интервал до 2-CdA, дни | 2-CdA-содержащие циклы терапии |
| 1 | 5 | LCH I | ПЗ | MP 30 мг/кг №3 – 2 блока | 81 | 2CdA 6 мг/м2/сут N5, AraC 200 мг/м2/сут N5 - 2 блока |
| 2 | 28 | LCH II | ПЗ | - | 37 | 2CdA 7 мг/м2/сут N5, DNR 45 мг/м2/сут N3; 2CdA 7 мг/м2/сут N5, AraC 1000 мг/м2/сут N5; 2CdA 7 мг/м2/сут N5, AraC 1000 мг/м2/сут N5 + Ida 8 мг/м2/сут N3 |
| 3 | 11,3 | LCH II | ПЗ | - | 46 | 2 CdA 8мг/м2/сут N5, AraC 1000 мг/м2/сут N5 – 4 блока |
| 4 | 6,3 | LCH II | ПЗ | - | 60 | 2 CdA 7 (9)мг/м2/сут N5, AraC 1000 мг/м2/сут N5 – 3 блока |
| 5 | 26 | LCH II | ПЗ | MP 20 мг/кг №3; Cy 500 мг/м2+Винкристин (Vcr) 0,05 мг/кг №4; Инфликсимаб 4 мг/кг №1; MTX 500 мг/м2/24 часа + МР 20 мг/кг №3 + 6-МП; MTX 500 мг/м2/24 часа; FLAG | 138 | 2 CdA 8(9)мг/м2/сут N5, AraC 1000 мг/м2/сут N5 – 3 блока (в 3 + Dauno 45 мг/м2/сут N2) |
| 6 | 15 | LCH II | ПЗ | Интенсивная фаза II (рукав В, Mtx+) | 87 | 2 CdA 6 мг/м2/сут N5, AraC 1000 мг/м2/сут N5 – 3 блока |
| 7 | 24 | LCH II | ПЗ | - | 29 | 2 CdA 6 мг/м2/сут N5, AraC 1000 мг/м2/сут N5 – 3 блока |
| 9 | 5,2 | LCH II | ЧО | VP16 100мг/м2 + МР 30 мг/кг №3 – 2 блока | - | - |
| 10 | 4 | LCH I | ПЗ | АТГ 25 мг/кг/курс; Cph 500 мг/м2 + DOXO 25 мг/м2; МР 30 мг/кг №3 | - | - |