
Молекулярно-генетическая диагностика и дифференцированная терапия гистиоцитарных пролиферативных заболеваний у детей 14. 01. 08 педиатрия 14. 01. 21 гематология и переливание крови
| Вид материала | Автореферат |
- Молекулярно-генетическая природа первичных гемофагоцитарных лимфогистиоцитозов в россии, 381.26kb.
- Аллогенная трансплантация гемопоэтических стволовых клеток в лечении врожденных и приобретенных, 1375.99kb.
- Факторы риска и контроль вирусных инфекций после трансплантации гемопоэтических стволовых, 767.07kb.
- Красняков Владимир Кириллович Совершенствование донорства крови и ее компонентов, 921.41kb.
- Прямое переливание крови (методические рекомендации), 154.15kb.
- Оптимизация инновационных технологий трансфузионного пособия пациентам регионального, 1245.79kb.
- Неопухолевые лимфаденопатии. 14. 00. 29 гематология и переливание крови, 1061.02kb.
- Обеспечение качества получения и клинического применения компонентов крови в субъекте, 404.49kb.
- кальцинированный аортальный стеноз-состояние системного гемостаза и реологических, 538.36kb.
- Иммуногематологическая оценка методов гемокомпонентной терапии у онкологических больных, 340.82kb.
Токсичность терапии с использованием 2-хлор-дезоксиаденозина.
Всего 15 пациентов получили 36 курсов комбинированной терапии 2-CdA+AraC, 9 пациентов получили терапию I линии, 6 пациентов – II линии. Все курсы комбинированной терапии сопровождались развитием анемии, тромбоцитопении и гранулоцитопении IV степени по шкале токсичности NCI CTC. Медиана длительности гранулоцитопении составила 12 (9-24) дней, медиана числа трансфузий тромбоконцентрата за весь период терапии составила 30 (13-52), медиана числа трансфузий эритроцитной массы составила 15 (8-23). Инфекционные осложнения включали фебрильную нейтропению после 34 из 36 (94%) курсов.
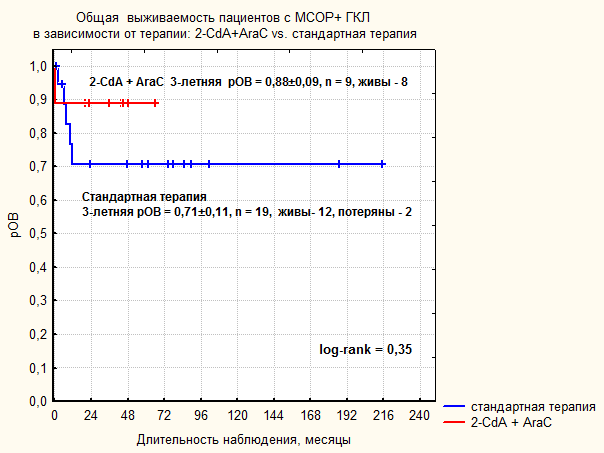 | 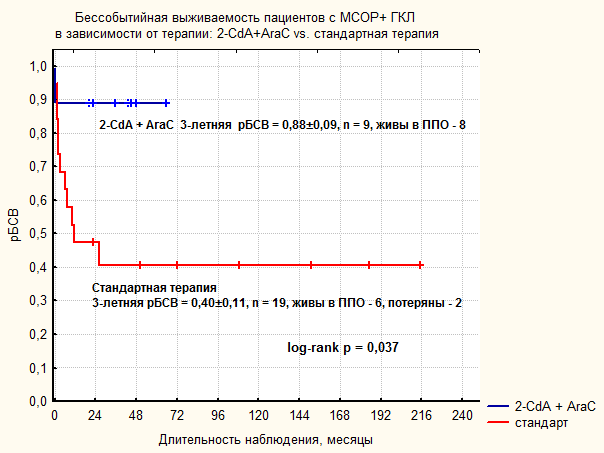 |
| Рисунок 24. Сравнительный анализ общей и бессобытийной выживаемости в группе МСОР+ ГКЛ в зависимости от терапии первой линии. | |
Микробиологически документированные инфекции включали 2 эпизода энтероколита с выявлением антигена C. difficile; 3 эпизода локальной реактивации BCG (M.bovis) c регионарным лимфаденитом; бактериемии с высевами: Sphingomonas pauzimobillis у 2 пациентов, C.parapsilosis, C.guilliermondii; инфекции мягких тканей с высевом C.parapsilosis, P.aeruginosae, A.baumanii, Staphylococcus spp, Enterococcus spp; респираторные вирусные инфекции с выявлением риновируса, аденовируса, метапневмовируса и парагриппа. При проведении сравнительного анализа в группе пациентов, получивших терапию 2-CdA+AraC, была определена достоверно большая частота развития инфекционных эпизодов (ИЭ) (р=0,039). При анализе структуры ИЭ в группе пациентов с 2-CdA показано достоверно более частое развитие инфекций мягких тканей (р=0,044) и документированных вирусных инфекций (р=0,016). Четырнадцать пациентов получили 74 курса монотерапии 2-CdA. Ни один курс монотерапии 2-CdA не сопровождался развитием нейтропении > II степени, тромбоцитопении > II степени, анемии > II степени. Случаев развития значимой органной токсичности после применения 2-CdA не зарегистрировано.
Обсуждение
Этиология ГКЛ остается предметом активных исследований. После демонстрации клональной природы патологических гистиоцитов в очагах ГКЛ (C.Wilman, 1994) установилось представление о ГКЛ как о клональном пролиферативном заболевании, гистогенетически связанной с эпидермальной клеткой Лангерганса. Попытки получить дополнительные доказательства злокачественной природы ГКЛ не увенчались успехом: не были выявлены повторяющиеся хромосомные аномалии, не удалось доказать инактивацию генов-супрессоров опухоли, противоречивые данные получены относительно состояния механизмов апоптоза и регуляции клеточного цикла. Сложность исследования патогенеза ГКЛ усугубляется отсутствием клеточных линий и адекватной мышиной модели заболевания. G.Badalyan-Very и соавт. в 2010 г. выявили мутации V600E BRAF в биообразцах пациентов с ГКЛ, эти данные стали первым за долгое время указанием на возможную роль классических механизмов активации протоонкогенов в развитии ГКЛ. Авторам исследования не удалось установить значимых клинических корреляций с наличием мутации V600E BRAF. Данные нашего исследования подтверждают, что мутация V600E BRAF выявляется в клетках патологического инфильтрата у значительной части пациентов с ГКЛ. Различия в доле BRAF-позитивных образцов между нашим исследованием и опубликованными данными обусловлены, возможно, недостаточной чувствительностью методики для выявления небольшой популяции ПКЛ на фоне нормальных клеточных элементов. В терапии пациентов с вовлечением «органов риска» результаты стандартной терапии первой линии, основанной на комбинации Vbl с Prn, неудовлетворительны. Вероятность рБСВ составила 40±11%. Более 50% пациентов нуждались в назначении терапии второй линии. Частота развития перманентных осложнений составила 63,6 %. Пять пациентов (29%) умерли от прогрессии заболевания. В работе показана высокая эффективность комбинированной ВХТ препаратами 2-CdA и AraC в лечении пациентов с ГКЛ, рефрактерных к стандартной терапии. Полный ответ был достигнут у 5 из 6 пациентов с рефрактерным течением ГКЛ, 5-летняя рОВ составила 66±15%, что является существенным прогрессом в сравнении с историческими данными, согласно которым рОВ у пациентов, не ответивших на терапию первой линии, не превышает 20%. Результаты пилотного исследования показали, что комбинация 2-CdA и AraC является наиболее эффективной медикаментозной терапией с точки зрения контроля ГКЛ. Полный продолжительный ответ был достигнут у всех пациентов, подлежавших оценке. Помимо высокой частоты ответа обращает внимание отсутствие случаев реактивации заболевания и перманентных осложнений, включая такое частое, как НД. Токсичность терапии 2-CdA+AraC обусловлена миелосупрессией и иммуносупрессией, тяжесть которых диктует высокие требования к сопроводительной терапии, в первую очередь – к качеству трансфузионной поддержки, профилактики и терапии инфекций. Этиологическая структура инфекций у пациентов, получивших терапию 2-CdA+AraC, отражает глубокий дефицит клеточного иммунитета и включает необычные для изолированной нейтропении патогены, такие как M.bovis, цитомегаловирус, респираторные вирусы и др. Таким образом, высоко эффективная терапия 2-CdA+AraC сопряжена с риском жизнеугрожающих осложнений. Окончательное место 2-CdA и AraC в терапии ГКЛ еще предстоит установить. Безусловно, в лечении рефрактерных форм МСОР+ ГКЛ у детей данная комбинация должна стать стандартом терапии. Возможность более раннего применения данной терапии, вероятно, будет определяться по мере выявления надежных биологических маркеров, коррелирующих с тяжестью течения ГКЛ, и позволяющих на этапе диагностики идентифицировать подгруппу пациентов, нуждающихся в наиболее агрессивной терапии. В любом случае, безопасное проведение такой терапии возможно только в центрах, обладающих опытом сопроводительной терапии пациентов с ОМЛ и пациентов после ТГСК.
Выводы
- В основе развития гистиоцитарных пролиферативных заболеваний (гистиоцитозов) лежат врожденные (ПГЛГ, ЮММЛ) либо приобретенные (ЮММЛ, ГКЛ) генетические дефекты, обусловливающие аномальную регуляцию пролиферации и функциональной активности гистиоцитов и их костномозговых предшественников. Идентификация молекулярных дефектов и выявление клинико-генетической корреляции необходимы для создания биологической классификации гистиоцитозов, разработки современного алгоритма диагностики, дифференциальной диагностики и терапии гистиоцитарных болезней.
- Молекулярно-генетический анализ генов PRF1, UNC13D, STX, STXBP, SH2D1A, XIAP и RAB27 показал, что доминирующее положение в генетической структуре ПГЛГ в репрезентативной выборке российских пациентов занимает FHL3 – вариант, обусловленный мутациями в гене UNC13D. Доля мутаций UNC13D составила 54% от всех пациентов с ПГЛГ, PRF1 и STX – по 3,5%. При анализе выявлены 9 новых и 4 ранее описанных мутации UNC13D. Высокая частота данного генетического варианта частично обусловлена распространением в популяции российских пациентов двух мутаций: c.2346_2349del и c.3037insG вследствие «эффекта основателя». Для выявления трех наиболее распространенных в России мутаций UNC13D, c.1828insA, c.2346_2349del и c.3037insG составляющих 48% мутаций в гене UNC13D, разработана тест-система на основе мультиплексной ПЦР-ПДАФ.
- В исследовании не выявлена значимая корреляция основных клинических и лабораторных характеристик ПГЛГ с гентическим вариантом FHL3. Отсутствие значимой клинико-генетической корреляции не позволяет использовать исходную клиническую и лабораторную информацию для предсказания генетического варианта ПГЛГ и селекции мишеней для молекулярно-генетического исследования.
- Стандартная иммуносупрессивная терапия этопозидом, дексаметазоном и циклоспорином А позволяет установить временный контроль над патологической активацией иммунной системы у 78% пациентов, однако в отсутствие ТГСК не способна излечивать пациентов с ПГЛГ. Частота реактиваций заболевания составила 95%. 5-летняя рОВ в группе ПГЛГ составляет 37±14 % при выполнении ТГСК, 0±0% при выполнении только ИСТ, log-rank р = 0,0005. Неудачи ТГСК обусловлены высокой трансплантационной смертностью, рTRM = 54±15%, вероятной причиной которой является длительный интервал до ТГСК (медиана – 14,6 месяцев) и кумулятивная токсичность множественных реактиваций заболевания и терапии.
- В группе пациентов с ЮММЛ наиболее распространенным соматическим генетическим дефектом являются мутации в гене PTPN11 (35%), c меньшей частотой встречаются мутации в генах NRAS (14%), CBL (13%) и KRAS (11%). Прямое секвенирование экзонов 3 и 13 PTPN11, 2 и 3 NRAS и KRAS, 8 и 9 CBL позволяет идентифицировать генетический дефект и верифицировать диагноз у 70% пациентов с ЮММЛ. Значимые отличия в клинико-лабораторной презентации генетических субвариантов ЮММЛ выявлены у пациентов с мутациями RAS в сравнении с PTPN11. Возраст манифестации заболевания составил 6,7 (RAS) и 24 (PTPN11) месяца, р = 0.0252. Содержание HbF 4,6 % (RAS) и 21% (PTPN11), р = 0.0008.
- Дифференцировочная терапия 13-RA + AraC индуцирует клинически значимый ответ у части пациентов с ЮММЛ. Ответ на ДТ коррелирует с ранним возрастом манифестации, низким содержанием HbF и мутациями в генах RAS и CBL. Пациенты, ответившие на ДТ, составили группу с лучшим показателем общей выживаемости, рОВ = 75±15%, как в сравнении с пациентами, не ответившими на ДТ, рОВ = 9±8%, так и в сравнении с пациентами, получившими ТГСК, рОВ = 0,38±13%. Ответ на ДТ не является эквивалентом излечения ЮММЛ, так как в гемопоэтическом компартменте персистирует мутантный клон и сохраняется риск развития злокачественных (Т-ОЛЛ, n - 1) и аутоиммунных болезней крови (ТТП, n – 1). ВХТ способна индуцировать кратковременный клинически значимый ответ у 50% пациентов, но не обеспечивает долгосрочную выживаемость.
- ТГСК является единственным методом терапии, способным излечивать ЮММЛ. При использовании миелоаблативных режимов кондиционирования рОВ и рБСВ в группе пациентов, получивших ТГСК, составляет 38±13% и 40±12%. Неудачи ТГСК обусловлены высокой трансплантационной смертностью, pTRM = 28±15%, и высокой частотой рецидивов ЮММЛ, 34±12%. Аллогенная ТГСК является терапией выбора для пациентов с ЮММЛ, не ответивших на ДТ, независимо от типа донора.
- Мутация V600E BRAF выявлена в патогистологических образцах 24% пациентов с ГКЛ. Значимой корреляции основных клинических и лабораторных показателей с мутационным статусом BRAF не выявлено. Мутация V600E BRAF является потенциальным маркером для диагностики и вероятной мишенью для терапии у части пациентов с ГКЛ.
- Комбинированная химиотерапия 2-Сda и AraC является эффективным методом лечения мультисистемного ГКЛ, резистентного к стандартной терапии, рОВ = 66±19%. Показано, что применение 2-Сda и AraC в терапии первой линии МСОР+ ГКЛ более эффективно в сравнении со стандартной терапией. Частота достижения полных ответов составила 100% vs. 33%, бессобытийная выживаемость, рБСВ = 88±9% vs 40±11%, частота формирования перманентных осложнений, 66% vs 0%. Применение ВХТ 2-Сda + AraC ассоциировано с выраженной миелосупрессией и иммуносупрессией и высоким риском развития инфекционных осложнений.
Практические рекомендации
- Для повышения эффективности лабораторной диагностики ПГЛГ в России необходимо этапное выполнение молекулярно-генетического анализа генов, ассоциированных с клиническим фенотипом ПГЛГ. На первом этапе целесообразно использовать тест-систему для выявления наиболее распространенных в российской популяции мутаций в гене UNC13D. На втором этапе – прямое секвенирование всех экзонов и экзон-интронных соединений гена UNC13D. На третьем этапе – прямое секвенирование генов PRF1, STX11, STXBP, RAB27 и, у пациентов мужского пола, SH2D1A, XIAP. Внедрение методики проточной цитометрии в лабораторную диагностику ПГЛГ позволит существенно оптимизировать затраты на генотипирование.
- Клинический диагноз ПГЛГ является абсолютным показанием к началу иммуносупрессивной (химио)терапии дексаметазоном и этопозидом, независимо от молекулярно-генетической верификации диагноза. Поиск неродственного донора следует инициировть в момент начала ИСТ. Алло ТГСК должна быть выполнена всем пациентам с верифицированным диагнозом ПГЛГ не позднее 4 месяцев от начала ИСТ. Выполнение ТГСК не должно быть ограничено наличием гистосовместимого донора и статусом ремиссии ГЛГ.
- Прямое секвенирование экзонов 3 и 13 PTPN11, 2 и 3 NRAS и KRAS, 8 и 9 CBL необходимо включить в ряд базовых диагностических исследований у пациентов с клиническим диагнозом ЮММЛ. Эффективность исследования может быть повышена путем поэтапного анализа в зависимости от исходных клинических и лабораторных характеристик. В группе пациентов младше 1 года с содержанием HbF < 10% целесообразно на первом этапе исследовать гены NRAS и KRAS, у пациентов старше 1 года с содержанием HbF > 10% - ген PTPN11.
- С целью определения тактики терапии пациентов с ЮММЛ целесообразно использовать клиническую стратификацию в соответствии с предложенной балльной шкалой, на основании трех показателей: возраста манифестации, содержания HbF и генетического дефекта.
- В группе пациентов младше 1 года с содержанием HbF < 10% и мутациями в генах NRAS и KRAS наиболее эффективным и безопасным методом терапии является дифференцировочная терапия 13-RA и низкими дозами AraC. ДТ индуцирует длительный клинико-гематологический ответ, у части пациентов – не требующий медикаментозной поддерживающей терапии. Пациенты, ответившие на ДТ, должны оставаться под наблюдением гематолога бессрочно в связи с риском развития поздних злокачественных и аутоиммунных болезней крови.
- Всем пациентам группы высокого риска и пациентам группы низкого и промежуточного риска, не ответившим на ДТ, должна быть выполнена ТГСК не позднее 6 месяцев от установления диагноза. Приоритетным источником ГСК является родственный или неродственный совместимый донор. Выполнение ТГСК не должно быть ограничено наличием гистосовместимого донора. В качестве иммуносупрессивного компонента кондиционирования целесообразно применение Flu. Целесообразно и оправдано этически включение всех пациентов с ЮММЛ, подлежащих ТГСК, в клинические исследования II-III фазы.
- Необходимо продолжить исследование роли мутации V600E BRAF и иных вероятных механизмов активации сигнального пути ERK/MAPK в патогенезе ГКЛ с использованием методик микродиссекции и молекулярного анализа на уровне одной клетки. Клиническое значение данной молекулярной аномалии должно быть исследовано проспективно.
- В терапии пациентов с ГКЛ, рефрактерных к стандартной ХТ, необходимо раннее (через 6-12 недель) использование комбинированной химиотерапии 2-СdA и AraC. Комбинированная терапия 2-СdA и AraC в терапии первой линии может применяться в рамках клинического исследования как эффективная альтернативой стандартной терапии у пациентов с МСОР+ ГКЛ. Проведение данной терапии должно быть ограничено стационарами, обладающими опытом ведения пациентов с ОМЛ и безусловным доступом к современной сопроводительной терапии.
Список работ, опубликованных по теме диссертации
- Пашанов Е.Д, Балашов Д.Н., Скоробогатова Е.В, Шипицына И.П., Трахтман П.Е., Дышлевая З.М., Скворцова Ю.В., Благонравова О.Л., Масчан М.А., Курникова Е.Е., Персиянцева М.И., Митюшкина Т.А., Масчан А.А., Румянцев А.Г. Характеристика инфекционных заболеваний у детей после трансплантации гемопоэтических стволовых клеток. Вопросы гематологии/ онкологии и иммунопатологии в педиатрии. 2005; 2: 68-81.
- О.В.Горонкова, А.А.Масчан, Е.В.Сунцова, Л.И. Жарикова, Г.Г.Солопова, Д.Д.Байдильдина, Л.А.Хачатрян, М.А.Масчан, Д.Н.Балашов, О.В.Макарова. «Эффективность и безопасность вориконазола в лечении инвазивных грибковых инфекций и эмпирической терапии фебрильной нейтропении у детей с онкогематологическими заболеваниями». Вопросы гематологии\онкологии и иммунопатологии в педиатрии, 2005, том 4, №3: с.87-94
- M.Maschan, G.Novichkova, E.Suntsova, L.Zharikova, O.Goronkova, D.Baidildina, L.Khachatryan, and A.Maschan 2-Chlordeoxyadenosine and intermediate-dose cytosine arabinoside combination therapy for high-risk langerhans cell histiocytosis Pediatric Blood & Cancer 2006.-V.46 . - Issue 3 . стр. 392– 405.
- Скоробогатова Е.В., Балашов Д.Н., Дышлевая З.М., Трахтман П.Е., Шелихова Л.Н., Скворцова Ю.В., Шипицына И.П., Курникова Е.Е., Пашко Ю.В., Благонравова О.Л., Персианцева М.И., Масчан М.А., Литвинов Д.В., Мякова Н.В., Бологов А.А., Масчан А.А., Румянцев А.Г.. Результаты трансплантации гемопоэтических стволовых клеток у детей. Опыт Федерального научно-клинического центра детской гематологии, онкологии и иммунологии на базе Российской детской клинической больницы. Вопросы гематологии/онкологии и иммунопатологии в педиатрии. 2006, т 6, №4, 29-38.
- Новичкова Г.А., Минков М., Масчан М.А., Чернов В.М. Гл. 45. Гистоиоцитозы В кн. Клиническая онкогематология. Под ред. проф. М.А. Волковой. - М.: Медицина. - 2007. - С. 891-912.
- Л.А.Хачатрян, Е.В.Самочатова, М.А.Масчан, Д.Д.Байдильдина, Г.Г.Солопова, А.А.Масчан. «Результаты терапии ювенильного миеломоноцитарного лейкоза у детей». Сборник материалов ХV Российского национального конгресса «Человек и лекарство»,Москва , 14-18 апреля 2008: с.423
- N.V.Poltavets, M.A.Maschan, A.V.Polyakov, G.A.Novichkova, A.A.Maschan Six new mutations in UNC13D gene in Russian patients with familial hemophagocytic lymphohistiocytosis (FHL) // Europ. J. Hum. Genet.– 2008. – V.16. – suppl 2. – p.247.
- Л.А.Хачатрян, М.А.Масчан, Е.В.Самочатова, М.М.Шнейдер, Д.Д. Байдильдина, Г.Г.Солопова, Е.В.Сунцова, Л.И.Жарикова, У.Н.Петрова, В.В.Синицына, Г.А.Новичкова, А.А.Масчан Дифференцировочная терапия с использованием 13-цис-ретиноевой кислоты и низких доз цитозин-арабинозида у детей с ювенильным миеломоноцитарным лейкозом. Онкогематолгия 2008, №1-2, стр. 34-38
- N.V.Poltavets, M.A.Maschan, A.V.Polyakov, G.A.Novichkova, A.A.Maschan Mutations in UNC13D gene are the most frequent cause of FHL in a group of Russian patients. Pediatric Blood &Cancer \\ 2009.- V.53.- Issue 4. стр.685–697.
- M.Maschan, G.Novichkova, D.Baidildina, L.Khachatryan, V.Sinizina, G.Solopova, U.Petrova, A.Maschan Up-front 2-Chlordeoxyadenosine-based combination chemotherapy in high-risk LCH: early results of pilot trial Pediatric Blood & Cancer 2009.-V. 53 .- Issue 4. стр. – 685-697.
- N.V.Poltavets, M.A.Maschan, I.G.Sermyagina, A.V.Polyakov, I.V.Kondratenko, A.A.Maschan, G.A.Novichkova Four SH2D1A mutations on 7 chromosomes detected in Russian patients with X-linked lymphoproliferative syndrome // Europ. J. Hum. Genet. - 2009. -V.17. - suppl 2. - стр.347.
- N.V.Poltavets, M.A.Maschan, I.G. Sermyagina, V.V. Zabnenkova, I.V.Kondratenko,G.A. Novichkova, A.A.Maschan, A.V.Polyakov Five SH2D1A mutations on 7 chromosomes detected in russian patients with X-linked lymphoproliferative syndrome (XLP) Сборник материалов 25-й конференции международной группы по изучению гистиоцитозов (The Histiocyte society), 2009 год, стр.43.
- M.A.Maschan, N.V. Poltavets, I.G. Sermyagina, V.V. Zabnenkova, I.V.Kondratenko, G.A. Novichkova, A.A.Maschan, A.V.Polyakov RAB27 mutations not detected in russian patients with familial hemophagocytic lymphohistiocytosis and X-linked lymphoproliferative syndrome. Сборник материалов 25-й конференции международной группы по изучению гистиоцитозов (The Histiocyte society), 2009 год, стр. 57.
- G.Solopova, D.Baidildina, E.Suntsova, O.Goronkova, U.Petrova, I.Kalinina, L.Khachatryan, V.Sinizina, G.Novichkova, A.Maschan, M.Maschan Front-line therapy of high-risk Langerhans cell histiocytosis with 2-Chlordeoxyadenosine and cytosine arabinoside: an update of a single center experience Pediatric Blood & Cancer 2010.-V. 55 .- Issue 5. стр. 880.
- N.V.Poltavets, M.A.Maschan, A.V.Polyakov, G.A.Novichkova, A.A.Maschan STXBP2 mutations are not detected in group of Russian patients with Familial hemophagocytic lymphohistiocytosis (FHL) // Europ. J. Hum. Genet.– 2010 - V.18. – suppl.1. - p.317.
- Полтавец Н.В., Масчан М.А., Поляков А.В., Масчан А.А., Новичкова Г.А. (2010) Исследование молекулярно-генетической природы семейного гемофагоцитарного лимфогистиоцитоза (FHL) в группе Российских больных // Молекулярная генетика – 2010 - № 2. - стр.143.
- М.А.Масчан, Г.А.Новичкова Гемофагоцитарный лимфогистиоцитоз Вопросы современной педиатрии 2009, том 8, №3, стр. 66-75
- Г.Г. Солопова, Д.Д. Байдильдина, Л.И. Жарикова, И.И. Калинина, У.Н. Петрова, Е.В. Сунцова, О.В. Горонкова, Л.А. Хачатрян, В.В. Синицина, Г.А. Новичкова, А.А.Масчан, М.А.Масчан Применение 2-хлордезоксиаденозина в терапии гистиоцитоза из клеток Лангерганса у детей Онкогематолгия 2010 год №3, стр. 8-15
- Полтавец Н.В., Масчан М.А., Масчан А.А., Новичкова Г.А., Поляков А.В. (2010) Мутации в гене UNC13D – наиболее частая причина семейного гемофагоцитарного лимфогистиоцитоза в группе российских больных Медицинская генетика, 2010, №3, стр.26-33
- Grunewald TG, Damke L,