
Синтез и биологическая активность карбо- и гетероциклов на основе тетрацианоэтилена 15. 00. 02 Фармацевтическая химия, фармакогнозия
| Вид материала | Автореферат |
- Синтез соединений на основе химических превращений производных α- оксокарбоновых кислот, 643.93kb.
- Синтез, свойства и биологическая активность енаминоамидов ацилпировиноградных кислот, 439.81kb.
- Синтез, свойства, биологическая активность n-гетериламидов α-оксокислот и продуктов, 367.21kb.
- Синтез, химические свойства и биологическая активность 1,4-дизамещенных 5-арил-3-гидрокси-3-пирролин-2-онов, 667.95kb.
- Синтез, свойства и биологическая активность продуктов взаимодействия 1,2,4-триазолов, 669.98kb.
- Синтез и свойства новых производных 2 (1,2,4 триазолил 5 тио)уксусных кислот 15. 00., 240.9kb.
- Примерная программа рекомендуется для направления подготовки (специальности) 111801, 717.4kb.
- Исследование по стандартизации и разработке лекарственных средств на основе листьев, 475.51kb.
- Исследования по разработке и стандартизации комбинированного антимикробного и регенерирующего, 405.42kb.
- «Фармацевтическая химия» специальность – 111201 Ветеринарный врач специализация Ветеринарная, 77.44kb.
а синтезированы из циклогексанонов 2
б синтезированы из дигидроксициклогексанов 11е,ж
Смешивали ТЦЭ (0,01 моль) с кетонами 1 а-д (0,012 моль) в неабсолютированном диоксане (20 мл) и 3-4 каплями конц. HCl. Циклогексаноны 2 е-к синтезированы в безводных растворителях (диоксан, ацетонитрил) в аналогичных условиях, но с использованием меньших количеств катализатора – 1-2 капли конц. HCl. В случае необезвоженных растворителей при взаимодействии ТЦЭ с кетонами 1е-т образуются 5-гидрокси-7-оксо-6-азабицикло[3.2.1]октан-1,2,2-трикарбонитрилы 13 е-т.
Более высокую реакционную способность циклогексанонов 2 е-т в сравнении с циклогексанонами 2а-д можно объяснить влиянием стерических факторов. Из анализа радикалов R2 и R3 в соединениях 2 следует, что у более реакционноспособных циклогексанонов 2 е-т, R2=R3=H (2е-з,л-п), R2 или R3 = CH3 (2 и,к,р,т), и R2 = Br (2с). Более устойчивые к процессам гидратации карбонильной и гидролизу аксиальной цианогруппы, и, соответственно, менее реакционноспособные циклогексаноны 2 а-д имеют более объемные заместители R2 и R3. У них сумма R2 + R3 равна двум или трем атомам углерода, что, по-видимому, уменьшает взаимодействия С=О и CN групп в циклогексанонах 2, а также ОН и CN групп в циклогександиолах 11. Аномальная активация С=О и одной CN-группы в положениях 1,3 по отношению друг к другу в циклогексанонах 2 объясняется их сближенностью. По расчетам, для циклогексанона 2ж расстояние О=С···СN равно 2,8Å. Согласно данным РСА в соединении 11ж существуют две водородные связи. Первая - между атомом кислорода О(2) и водородом НО(1) ее длина 1,892Å. Вторая - между водородом аксиального гидроксила О(2)Н и азотом N(1) равная 2,005Å (рис. 2). Расстояние О(2) – С(7) равно 2,786Å. Эти факторы, по нашему мнению, способствуют легкому гидролизу цианогруппы циклогексанов 2,11.
Д
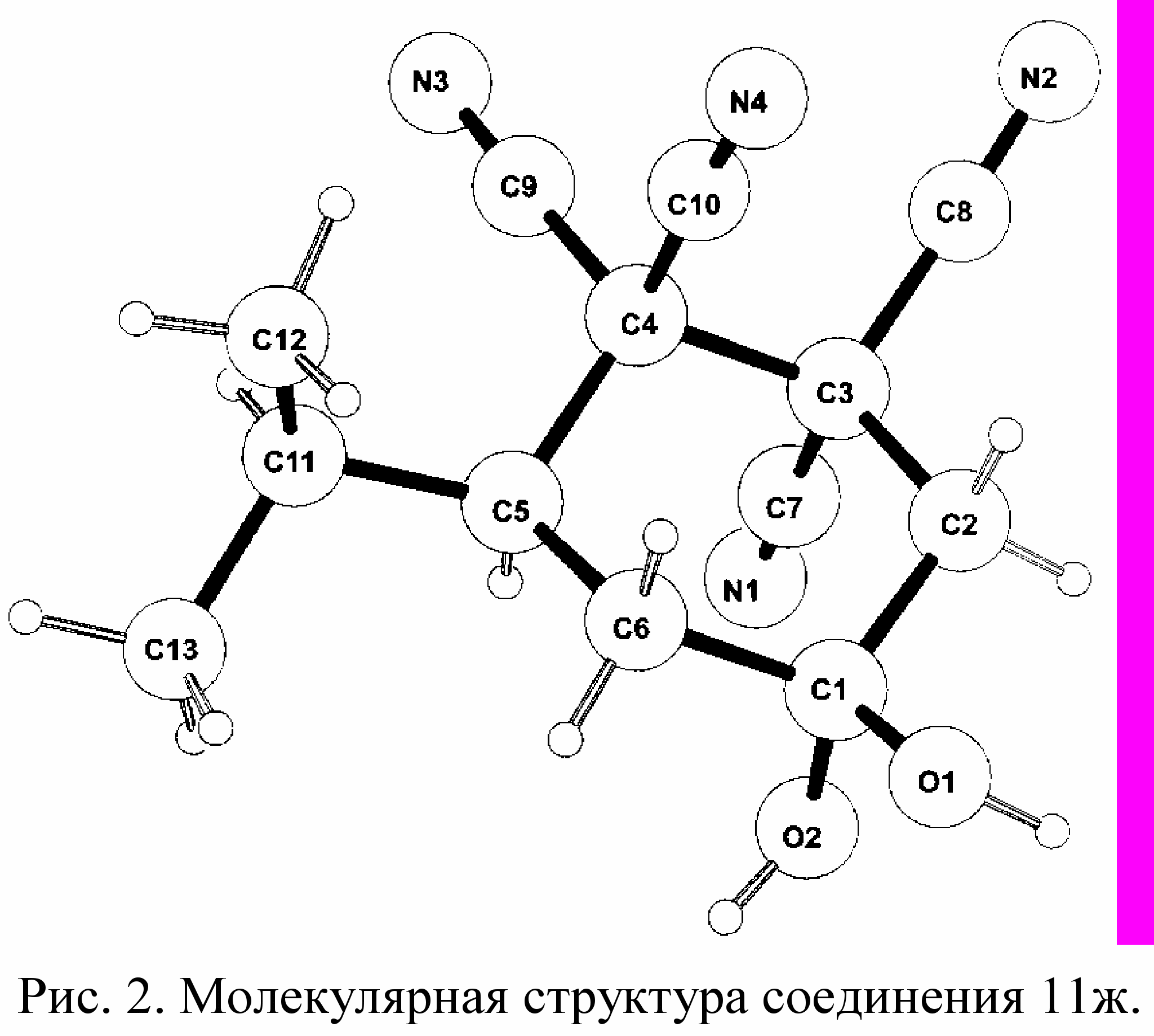
оказательством последовательности стадий показанных на схеме 6 служит выделение соединений 11 и 12. Диолы 11 е, ж были синтезированы при выдерживании циклогексанонов 2 е, ж в водном диоксане с каталитическим количеством соляной кислоты. Карбоксамиды 12е,ж получены нами при кипячении соединений 11е,ж в 2-пропаноле. Превращение 12→13 (схема 6) подтверждается переходом карбоксамидов 12е,ж в азабициклы 13е,ж при кипячении в диоксане с конц. HCl. Кроме того, соединения 13а-к синтезированы нами из циклогексанонов 2а-к при кипячении их в смеси диоксан – конц. HCl в соотношении 1:1.
- Реакции тетрацианоэтилена с галогенкетонами и
дикарбонильными соединениями
Исследования реакций ТЦЭ с α,β-непредельными кетонами показали, что 4-оксоалкан-1,1,2,2-тетракарбонитрилы, содержащие в положении 5 или 6 функциональную группу, способную реагировать с СН кислотным фрагментом, имеют тенденцию к формированию циклоалкана. Мы пришли к выводу, что для получения других производных тетрацианоциклоалканов необходимо изучить реакции ТЦЭ с α- и β-галогенкетонами и с некоторыми дикарбонильными соединениями у которых после тетрацианоэтилирования устанавливается благоприятная стерическая ситуация для (CN)2CH···O=C взаимодействия.
1.2.1. Взаимодействие тетрацианоэтилена с α-хлоркетонами
Мы разработали новый метод синтеза производных тетрацианоциклопропана – (2,2,3,3-тетрацианоциклопропил)кетонов 15, заключающийся во взаимодействии ТЦЭ с α-хлоркетонами, с помощью которого получены алкилциклопропилкетоны 15а-г, спиросоединения 15д,е и арилциклопропилкетоны 15ж-и с выходами 60-83%. Полагаем, что реакции начинаются тетрацианоэтилированием по α-галоидзамещенному фрагменту кетонов с образованием 3-хлор-4-оксоалкан-1,1,2,2-тетракарбонитрилов i3. Они неустойчивы и элиминируют HCl, образуя при этом циклопропилкетоны 15. Найденный нами способ получения тетрацианоциклопропанов 15, по сравнению с методом Видеквиста, имеет определенные преимущества. Еще в 1963 г Харт и Фриман сообщили, что при смешении некоторых кетонов: пинаколина, этилбутилкетона, диамилкетона, диизопропилкетона, бензофенона с броммалононитрилом в присутствии йодида калия тетрацианоциклопропаны с заместителями С(CH3)3, i- C3H7, C3H7, C4H9, C6H5 не образуются.
Схема 7.
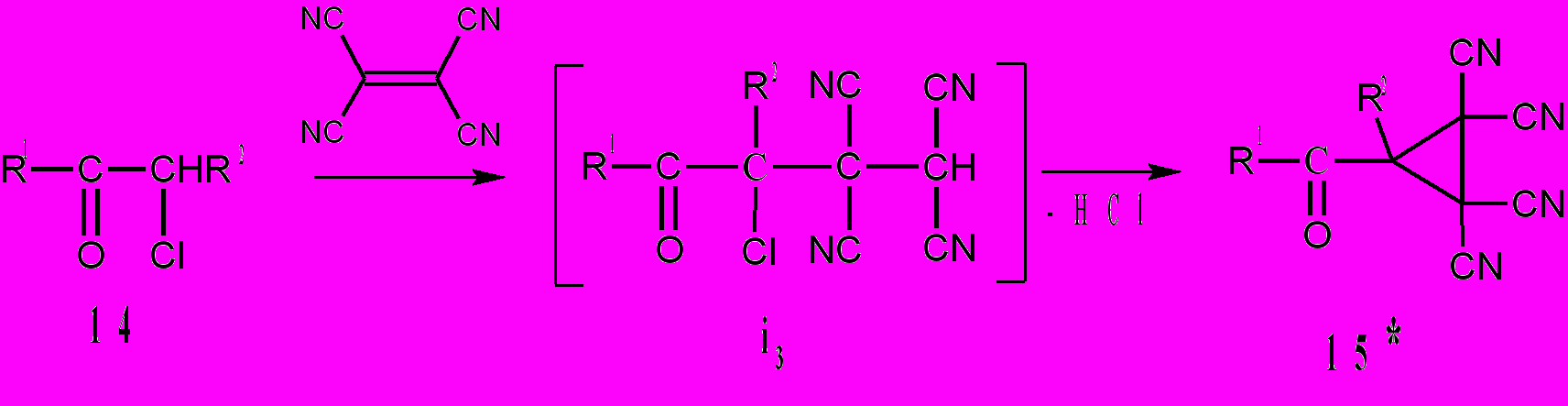
R1 = CH3, R2 = H (а); R1 = C(CH3)3, R2 = H (б); R1 = CH3, R2 = i-C3H7 (в); R1 = CH3, R2 = C3H7 (г); R1 + R2 = (CH2)3 (д)*; R1 + R2 = (CH2)4 (е); R1 = C6H5, R2 = H (ж); R1 = 4-CH3C6H4, R2 = H (з);R1 = 4-ClC6H4, R2 = H (и)
К
Рис. 3. Молекулярная структура
соединения 15д
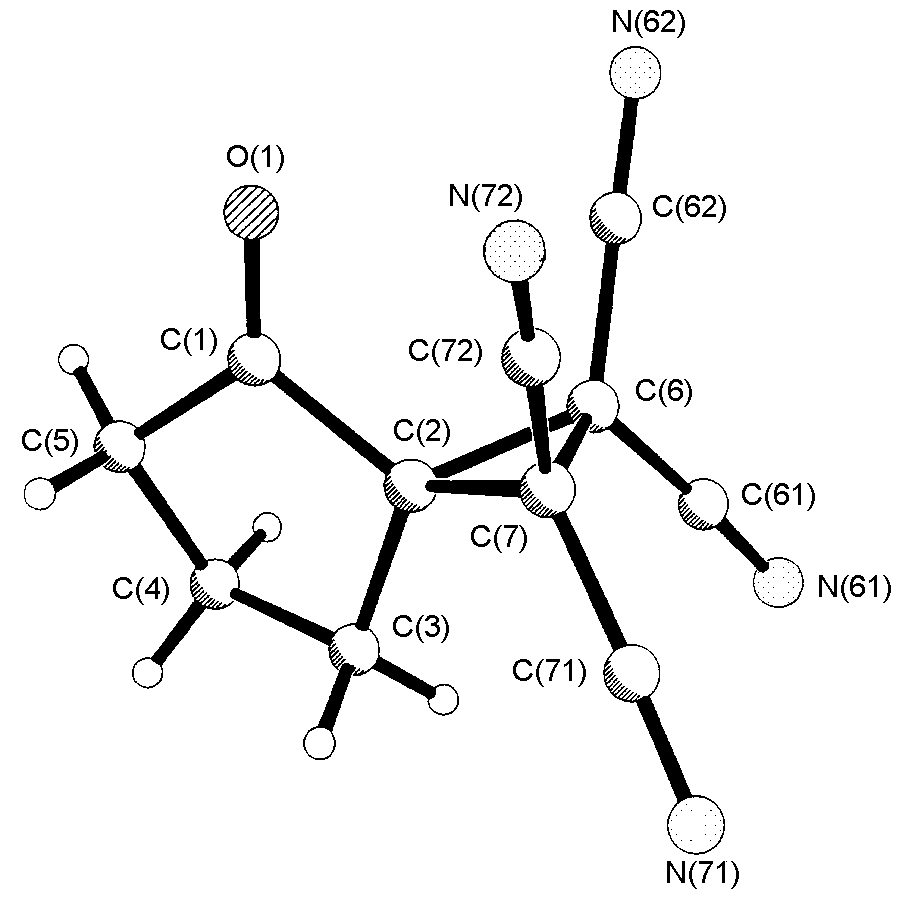
ак показано на схеме 7, наш метод позволяет синтезировать тетрацианоциклопропаны с этими заместите-лями. Из структурных особенностей циклопропанов 15 следует выделить копланарное расположение σ-связей фрагментов С(6)С(62)N(62) и С(7)С(72)N(72). Это является допол-нительным активирующим фактором CN-групп. Близкое расположение С=О и двух CN групп, ориентиро-ванных в одном направлении по отношению к циклопопановому кольцу является фактором наиболее вероятного протекания различных взаимодействий и превращений между ними. Однако, в условиях синтеза соединений 15 присутствие воды и каталитических количеств HCl к каким-либо превращениям циклопропанов не приводит. При переходе циклогексанона 2ж (Е=63,83 ккал/моль) в гидрат 11ж (Е=63,55 ккал/моль) (схема 6) происходит, хотя и незначительное, но уменьшение общей энергии. Гипотетический гидрат циклопропана 15д (Е=43 ккал/моль) энергетически менее выгоден в сравнении с его негидратированной формой 15д (Е=39,16 ккал/моль), что и является препятствием для протекания процессов аналогичным реакциям 11→i2→12→13 (схема 6).
1.2.2. Реакционная способность тетрацианоэтилена по отношению к β-бромкетонам
Наибольшее распространение в синтезе 1,1,2,2-тетрацианоциклобутанов имеет реакция [2+2] циклоприсоединения. Легче всего превращения данного типа протекают при взаимодействии ТЦЭ с алкенами, двойная связь которых активирована электронодонорными заместителями. Как правило, этим процессам приписывают механизм несогласованного циклоприсоединения, где образование циклобутанов происходит постадийно через цвиттер-ионный интермедиат. Принципиально новый подход к синтезу тетрацианоциклобутанов найден нами при изучении взаимодействия ТЦЭ с β-бромкетонами. Происходит быстрое (за 2-3 мин), с высокими выходами (91-96%), образование 1-ацетонил-2,2,3,3-тетрацианоциклобутанов 17. Кроме того, циклобутаны 17 можно получить реакцией ТЦЭ с α,β-непредельными кетонами 1ж,з,о в присутствии конц. НBr. Однако, в этом случае, их образование протекает дольше – за 12 ч, и со сравнительно низкими выходами (56-71%). Кето-енольная таутомерия бромкетона 16 затрагивает фрагмент CHBr. Дегидробромирование енольной формы кетона 16 приводит к интермедиату i4 c цепью сопряжения, включающей атом углерода при R1 и R2. Мы полагаем, что вместо обычного тетрацианоэтилирования исходных кетонов 1ж,з,о и 16 по С(О)СН3 фрагменту в этих случаях имеет место взаимодействие ТЦЭ с атомом углерода при R1 и R2 интермедиата i4, что приводит к образованию интермедиата i5. Последующие процессы миграции протона и циклизации дают циклобутаны 17. Дегидробромирование кетона 16 до β,γ-непредельного кетона и его взаимодействие с ТЦЭ не представляется реальным направлением превращения 16→17. Подтверждением этому служит дополнительно проведенная реакция ТЦЭ с метилаллилкетоном, не приводящая к каким-либо индивидуальным соединениям. Поэтому мы считаем, что дегидробромирование бромкетона 16 протекает через енольную форму.
Схема 8.
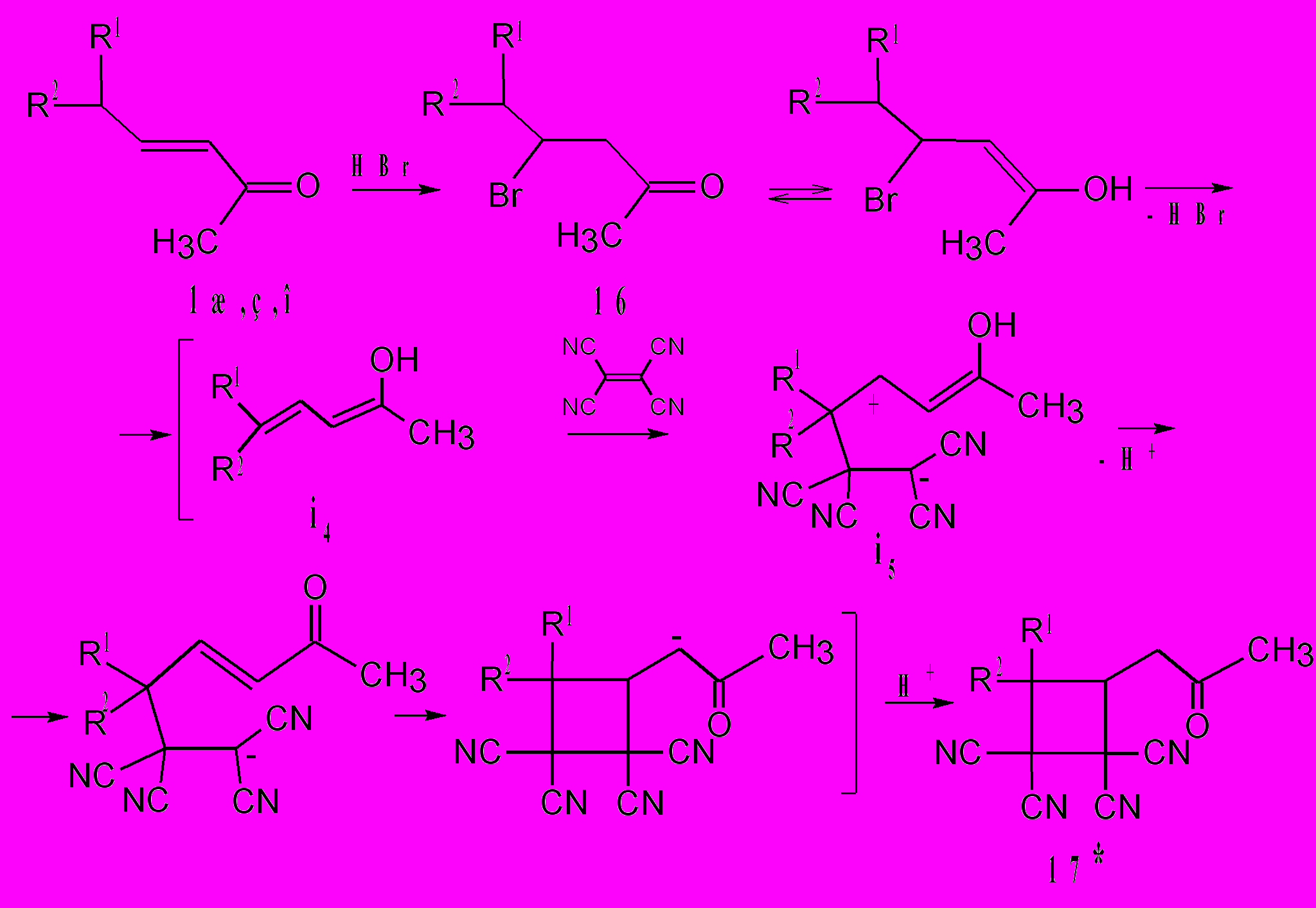
R1 = C2H5, R2 = H(а)*; R1 = R2 = СH3(б); R1 = C6H5, R2 = H(в)
Активность диена i4 к ТЦЭ, по схеме [2+2] циклоприсоединения, по-видимому, обусловлена эффектом сопряжения кратных связей и наличием электронодонорных групп. Об удобном методе синтеза циклобутанов реакцией сопряженных диенов, имеющих S-trans конформацию с ТЦЭ, сообщили Нишида С. и соавторы (1980). Процессы гидратации циклобутанов 17 в ходе их синтеза и дальнейшие НО···CN взаимодействия, как в циклогексанолах 11 не протекают вследствие увеличения энергии молекул. По расчетам, для циклобутана 17а Е=68,09 ккал/моль, а для его гидрата Е=76,25 ккал/моль.
- Взаимодействие тетрацианоэтилена с дикарбонильными
соединениями
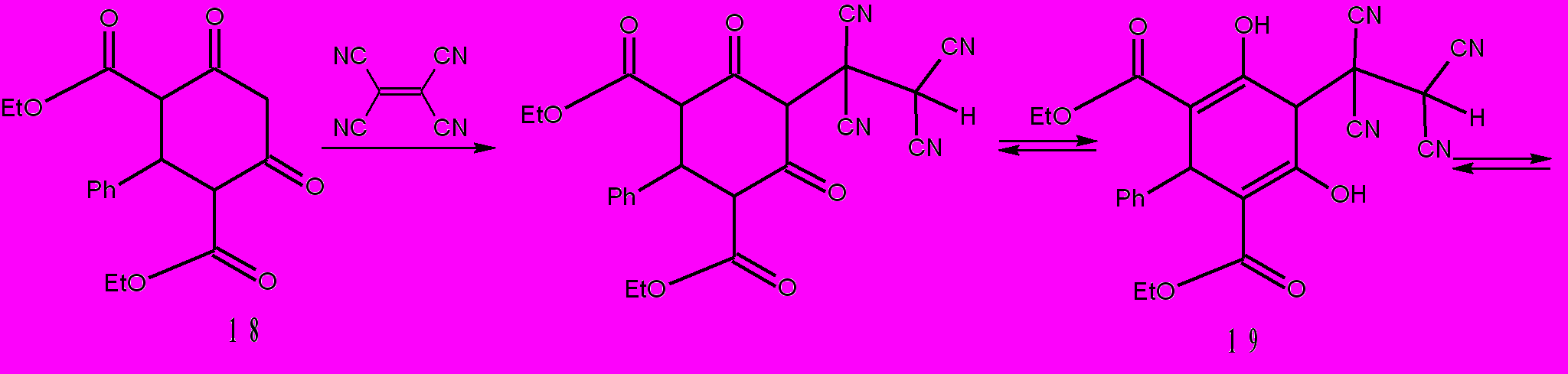
Известно, что ТЦЭ взаимодействует с β-дикарбонилными соединениями с образованием ОАТ, пирролов, пиранов и фуранов. Мягкие условия проведения процессов, высокие выходы, потенциальная биологическая активность аддуктов ТЦЭ и β-дикарбонильных соединений – обусловили повышенное внимание к этим объектам. Наиболее сложные превращения протекают при взаимодействии ТЦЭ с ацетилацетоном, ацетоуксусным эфиром и их производными в присутствии пиперидина в бензоле. Последовательность стадий для них выглядит следующим образом: ТЦЭ + β-дикарбонильное соединение → ОАТ → производные фурана → производные пиррола. В случае взаимодействия ТЦЭ с ацетилацетоном использование в качестве растворителя ацетонитрила изменяет ход процессов и образуется 4Н-пиран. Наиболее характерно образование 4Н-пиранов для реакций ТЦЭ с циклическими β-дикетонами (циклогександион-1,3, димедон, 4-гидроксикумарин). Из всего этого следует, что ОАТ, образующиеся при взаимодействии ТЦЭ и β-дикетонов, имеют тенденцию циклизоваться в реакционноспособные, полифункциональные гетероциклические соединения. В связи с вышеуказанным, представляло интерес изучение взаимодействия ТЦЭ с циклическим дикетоном 18, имеющим в своей структуре фрагмент ацетилацетона и ацетоуксусного эфира. Для дикетона 18 возможно несколько направлений реакции тетрацианоэтилирования: в положение 2 и в положение 4 или 6 цикла.
В первом случае должен был образовываться в качестве интермедиата или конечного продукта замещенный фуран или пиран, во втором должна была реализоваться циклизация (CN)2СН···C=О и формирование циклогексана. В действительности же тетрацианоэтилирование дикетона 18 идет в положение 2. Процесс протекает за 1-2 ч. Соединение 19 выделяется при разбавлении реакционной смеси водой. Если реакционную смесь в диоксане выдержать при комнатной температуре в течение 24 ч, то образуется хинолин 21. Соотношение ТЦЭ к катализатору (НCl) 1:0,1
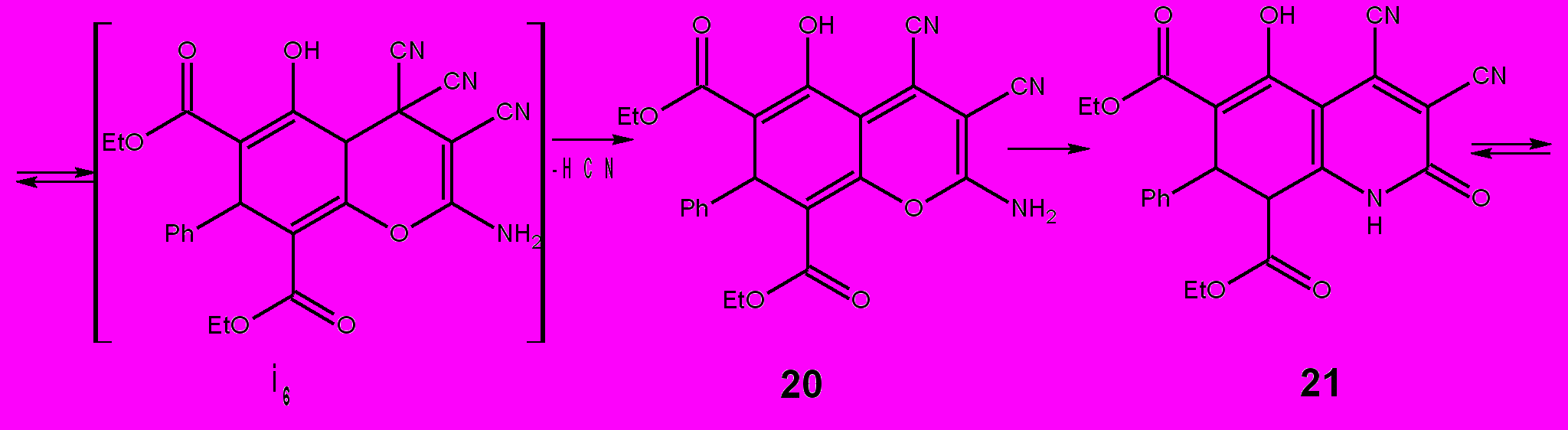
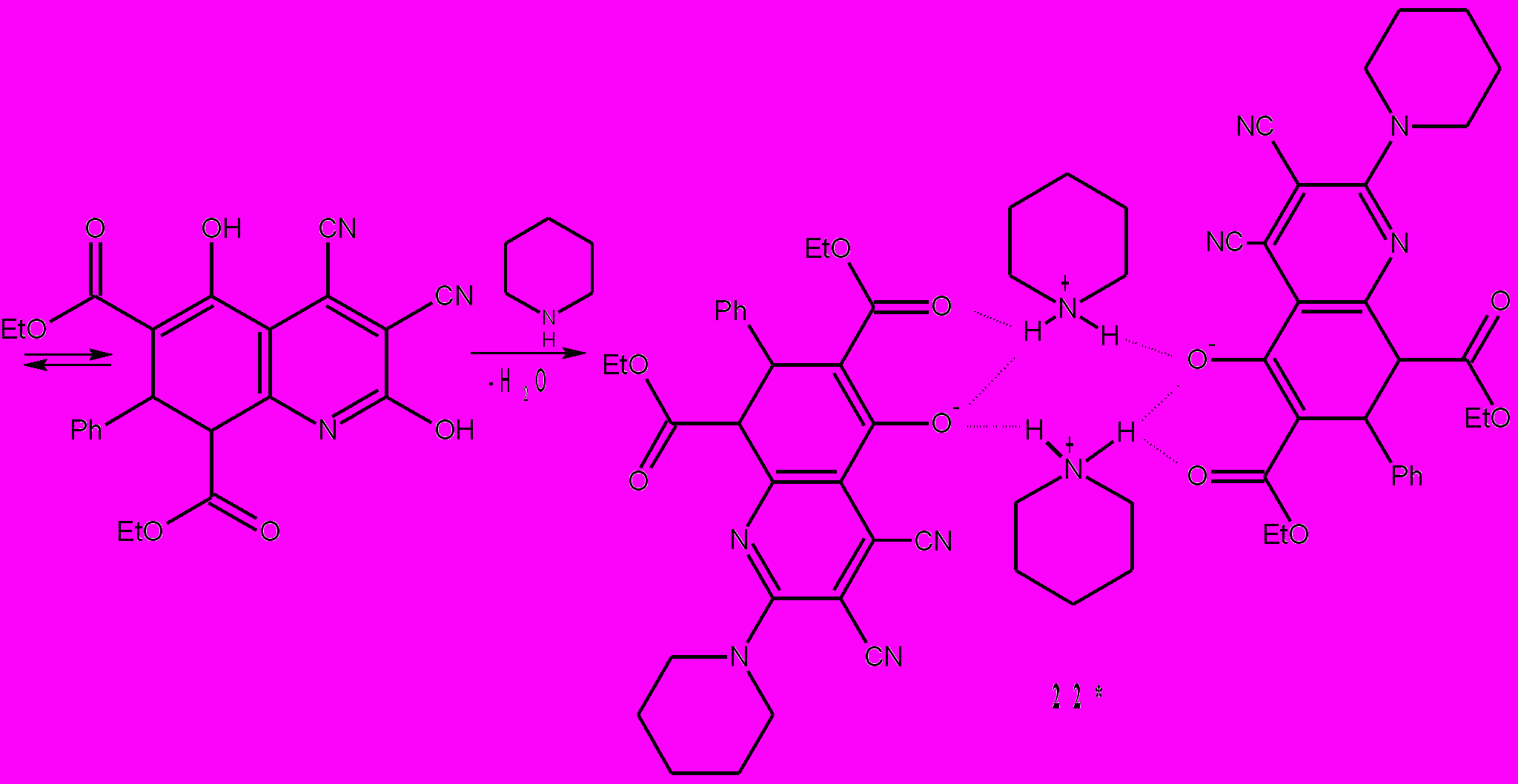
 0,2 (моль). Образование пиранового цикла при внутримолекулярном взаимодействии гидрокси- и цианогрупп является обратимым процессом и в определенных условиях пирановый цикл может раскрываться. Так, 5-Alk-2-амино-3-цианопиридины образуются при нагревании 5-Alk-2-амино-3-цианопиранов в уксусной кислоте в присутствии ацетата аммония. Вероятно, процессам образования соединения 20 и рециклизации пиран 20 → хинолин 21 способствуют наличие в них сопряженных кратных связей и карбэтоксильные группы. Так, например, димедон реагирует с ТЦЭ только с образованием пирана. Последовательность стадий 18→21 подтверждена нами выделением пирана 20 и осуществлением превращения пиран 20 → хинолин 21. Пиран 20 мы синтезировали из дикетона 18 и ТЦЭ в диоксане при соотношении ТЦЭ:НСl = 1:0,010,02 (моль) и выдерживании реакционной смеси при комнатной температуре в течении 2-3 ч. Условия проведения рециклизации пирана 20 до хинолина 21 аналогичны условиям проведения процесса ТЦЭ + дикетон 18 → соединение 21: растворитель диоксан, комнатная температура, время реакции 22-24 ч, соотношение ТЦЭ к HCl = 1:0,10,2 (моль). Уникальное сочетание функциональных групп придало соединению 21 необычную способность реагировать с пиперидином с трансформацией пиридонового цикла в пиридиновый и, кроме того, за счет избытка пиперидина образовывать устойчивый хорошо кристаллизующийся комплекс 22. Реакция протекает в этаноле с избытком пиперидина. Полагаем, что вначале происходит протонирование пиперидина и образование водородных связей O1-H, O1´-H´, O5-H, O5´-H´. Затем, вероятно, образуются σ-связи С2-N и C2´-N´.
0,2 (моль). Образование пиранового цикла при внутримолекулярном взаимодействии гидрокси- и цианогрупп является обратимым процессом и в определенных условиях пирановый цикл может раскрываться. Так, 5-Alk-2-амино-3-цианопиридины образуются при нагревании 5-Alk-2-амино-3-цианопиранов в уксусной кислоте в присутствии ацетата аммония. Вероятно, процессам образования соединения 20 и рециклизации пиран 20 → хинолин 21 способствуют наличие в них сопряженных кратных связей и карбэтоксильные группы. Так, например, димедон реагирует с ТЦЭ только с образованием пирана. Последовательность стадий 18→21 подтверждена нами выделением пирана 20 и осуществлением превращения пиран 20 → хинолин 21. Пиран 20 мы синтезировали из дикетона 18 и ТЦЭ в диоксане при соотношении ТЦЭ:НСl = 1:0,010,02 (моль) и выдерживании реакционной смеси при комнатной температуре в течении 2-3 ч. Условия проведения рециклизации пирана 20 до хинолина 21 аналогичны условиям проведения процесса ТЦЭ + дикетон 18 → соединение 21: растворитель диоксан, комнатная температура, время реакции 22-24 ч, соотношение ТЦЭ к HCl = 1:0,10,2 (моль). Уникальное сочетание функциональных групп придало соединению 21 необычную способность реагировать с пиперидином с трансформацией пиридонового цикла в пиридиновый и, кроме того, за счет избытка пиперидина образовывать устойчивый хорошо кристаллизующийся комплекс 22. Реакция протекает в этаноле с избытком пиперидина. Полагаем, что вначале происходит протонирование пиперидина и образование водородных связей O1-H, O1´-H´, O5-H, O5´-H´. Затем, вероятно, образуются σ-связи С2-N и C2´-N´.Схема 9.
Другого типа циклизации осуществлены нами при взаимодействии ТЦЭ с дикетонами 23 и 25. Так, ТЦЭ с 1,2-циклогександионом 23 и бис(циклогексанон-2-ил)метаном 25 реагирует с образованием соответственно: 6,6,7,7-тетрацианобицикло[3.2.1]октан-1,8,8-триола 24 и 7-имино-2-спиро-1΄-[2΄-оксоциклогексан]-4,5-тетраметилен-6-оксабицикло[3.2.1]ок-тан-1,8,8-трикарбонитрила 26. Основное отличие этих превращений (схемы 10,11) от реакций ТЦЭ с β-дикарбонильными соединениями – это внутримолекулярное взаимодействие кислотного фрагмента CH(CN)2 с карбонильной группой, приводящее к формированию циклоалканового кольца. При взаимодействии с дикетоном 23 происходит необычное 1,3-циклоприсоединение и образование устойчивого гидрата 24 с выходом 78%.
Схема 10.
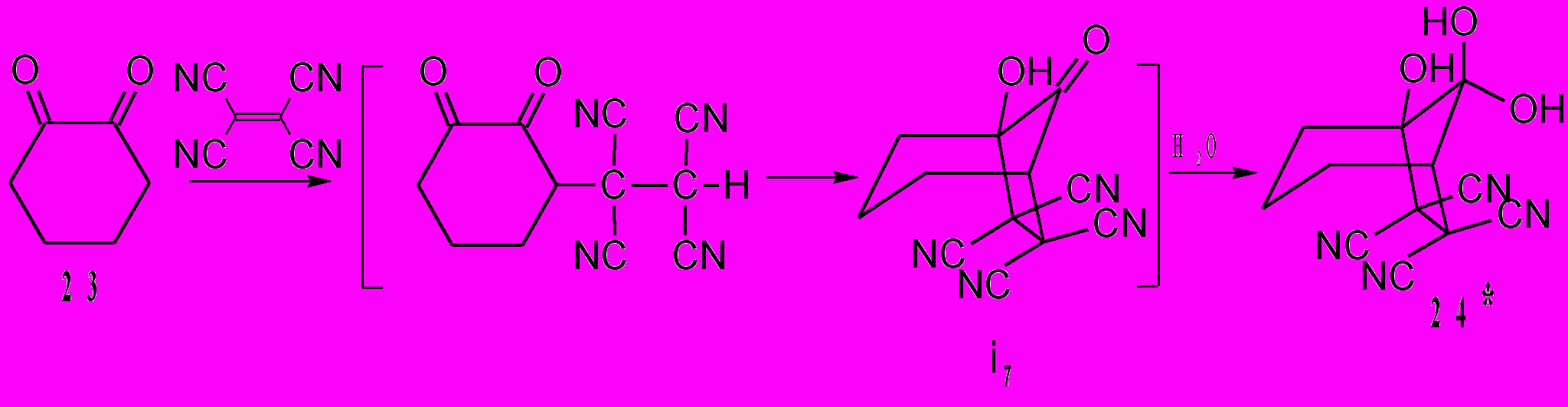
Вероятно, аналогично происходит взаимодействие ТЦЭ с дикетоном 25 – это тетрацианоэтилирование и формирование алканового цикла. Цикл, однако, в этом случае формируется не пяти-, как в реакции с дикетоном 23, а шестичленный. Следующее отличие от предыдущей реакции заключается в реализации НО···CN взаимодействия, приводящему к соединению 26.
Схема 11.
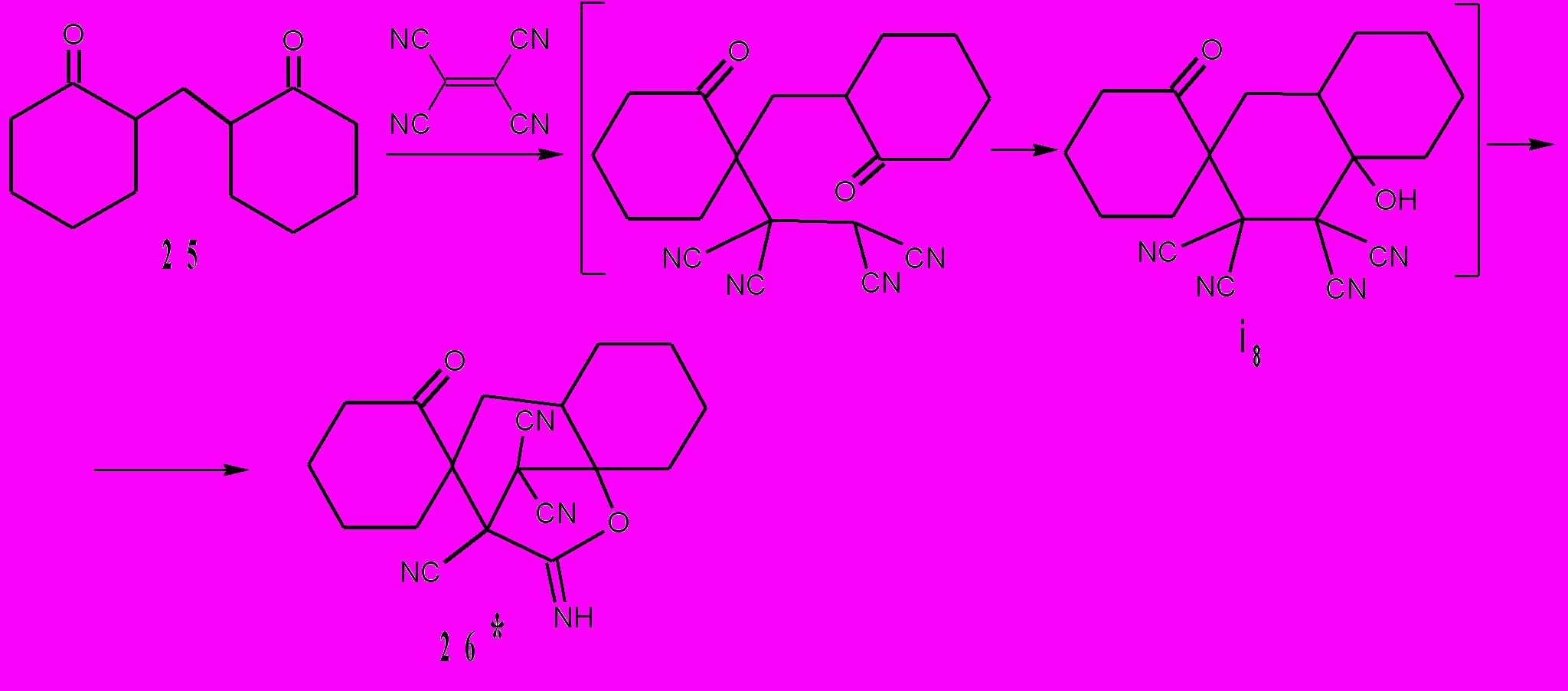
Взаимодействие ТЦЭ с карбонильными соединениями открывает путь к уникальным карбо- и гетероциклическим структурам, получение которых другими методами невозможно или затруднительно. Этот путь лежит через образование ОАТ. Из литературных данных (Шаранин Ю.А., 1998) следует, что аддукты Михаэля ТЦЭ и дикарбонильных соединений очень редко удается получить в индивидуальном состоянии, поскольку они высокореакционноспособны. Но их аналоги – ОАТ, образованные из ТЦЭ и монокарбонильных соединений почти всегда можно выделить из реакционных масс разбавлением водой или гексаном. Возможность выделения ОАТ создает условия для всестороннего исследования их свойств. Впервые тетрацианоэтилирование простейших кетонов (ацетон, циклогексанон, ацетофенон и их аналоги) проведено Миддлетоном (1957). Мы распространили эту реакцию на более сложные кетоны, в том числе природные и биологически активные, и провели тетрацианоэтилирование пинаколина, метил(1-адамантил)кетона, α- и β-иононов, циклододеканона, ментона, дигидроэпиандростерона. Таким образом, реакции ТЦЭ с карбонильными соединениями перспективны не только для синтеза новых карбо- и гетероциклов, но и при трансформировании и модификации обширных классов карбонильных соединений, имеющих фрагмент CR13C(O)CHR22.
1.3. Реакции 4-оксоалкан-1,1,2,2-тетракарбонитрилов с альдегидами
Анализируя различные реакции тетрацианоалканонов мы обнаружили, что наиболее подходящими реагентами для перевода их в более устойчивые и хорошо кристаллизующиеся циклические производные являются альдегиды. Они позволяют быстро, в мягких условиях и с высокими выходами превращать тетрацианоалканоны в карбо- и гетероциклы. Благодаря этому нам удалось вовлечь в эти реакции органические соединения сложного строения, включая природные и биологически активные. Ко всем классам синтезированных нами соединений, и особенно к бициклическим пиранам 29, был проявлен интерес со стороны Национального института рака NCI (National Cancer Institute, USA).
Из литературных данных известно, что пираны представляют интерес в качестве аналогов природных соединений, физиологически активных веществ, красителей и пестицидов. Взаимодействие тетрацианоалканонов 28 с альдегидами, не содержащими каких-либо функциональных групп, приводит к образованию биологически активных кислородосодержащих гетероциклов 29 пирановой структуры. Метод получения бициклических пиранов 29 прост: после завершения реакции тетрацианоэтилирования кетонов, альдегид загружается в реакционную смесь без выделения образующихся тетрацианоалканонов. При этом, по-видимому, образуются пираны i9, которые затем циклизуются в бициклы 29. При осуществлении превращений 27→29 без выделения тетрацианоалканонов 28 выходы соединений 29 составляют 85-92%. Реакции тетрацианоэтилированных кетонов 28 с альдегидами протекают за 20-30 мин. Диоксабициклы 29 выделяли разбавлением смеси водой. Соединения 29 представляют собой устойчивые при хранении и хорошо кристаллизующиеся из многих органических растворителей вещества. Ранее продуктам взаимодействия тетрацианоалканонов 28 с альдегидами приписывалась другая структура. Предполагалось, что циклизация интермедиата i9 до замещенного диоксабицикло[2.2.2]октана осуществляется в результате 1,4 взаимодействия групп ОН и СN. Сравнение энергии молекулы 29и (Е=55,25 ккал/моль) с энергией гипотетической структуры соответствующего замещенного диоксабицикло[2.2.2]октана (Е=64,19 ккал/моль) дает основание утверждать, что это взаимодействие не реализуется.
Схема 12.
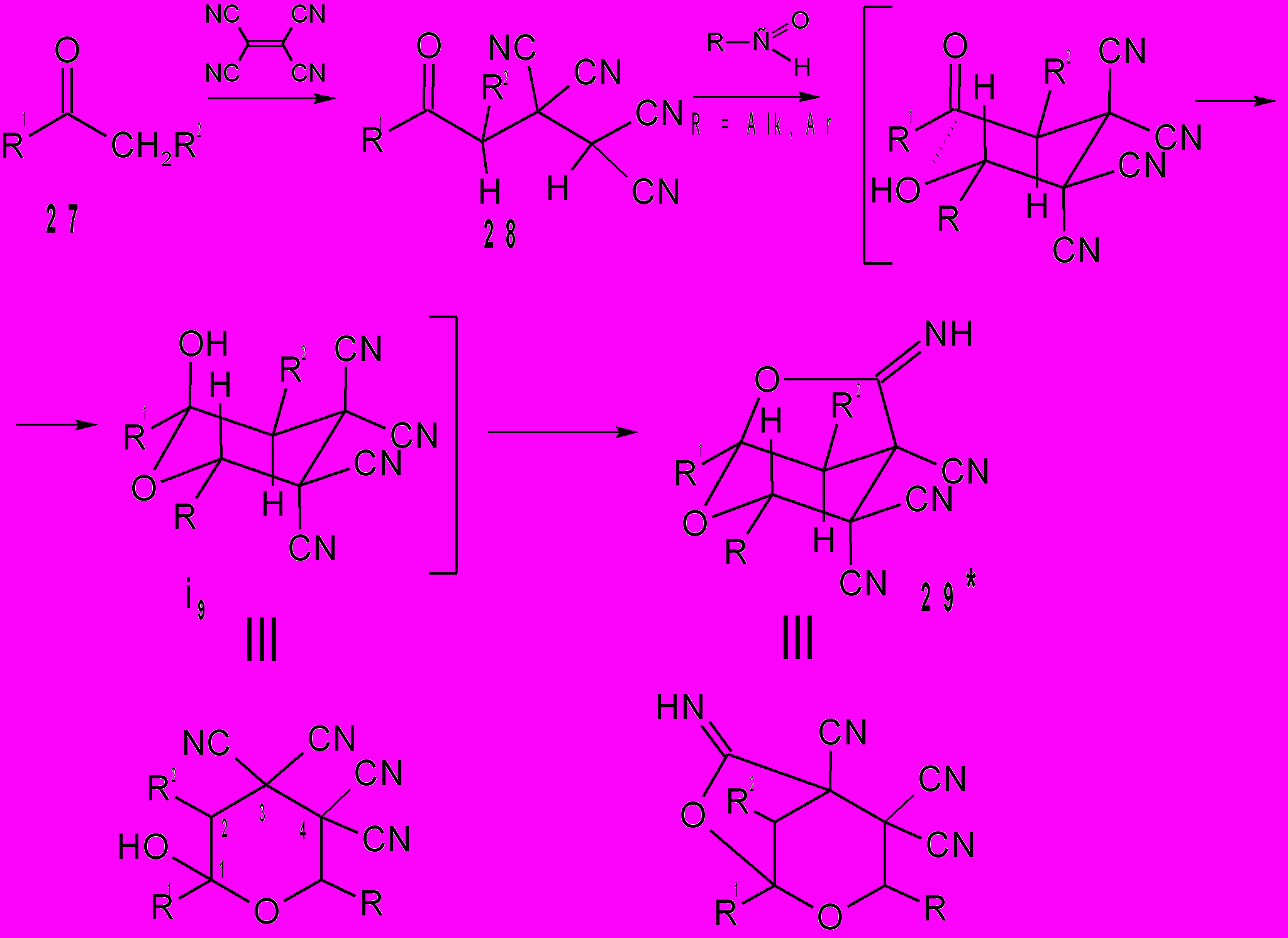
29: R1 + R2 = (CH2)4, R = 2-Fu (а); R1 + R2 = (CH2)4, R = i-C3H7(б); R1 + R2 = (CH2)4, R = CH3 (в); R1 + R2 = (CH2)4, R = H (г); R1 = R2 = CH3, R = 3-O2NC6H4 (д); R1 = R2 = CH3, R = C6H5 (е); R1 = R2 = CH3, R = 2-Fu (ж); R1 = R2 = CH3, R = i-C3H7 (з); R = R1 = R2 = CH3 (и); R1 = R2 = CH3, R = H (к); R1 = CH3, R2 = H, R = C2H5 (л); R1 = CH3, R2 = R = H (м); R1 + R2 = (CH2)4, R = C6H5 (н)*; R1 + R2 = (CH2)10, R = H (о);R1 + R2 = (CH2)10, R = C6H5 (п); R1 + R2 = (CH2)10, R = 4-Py (р).
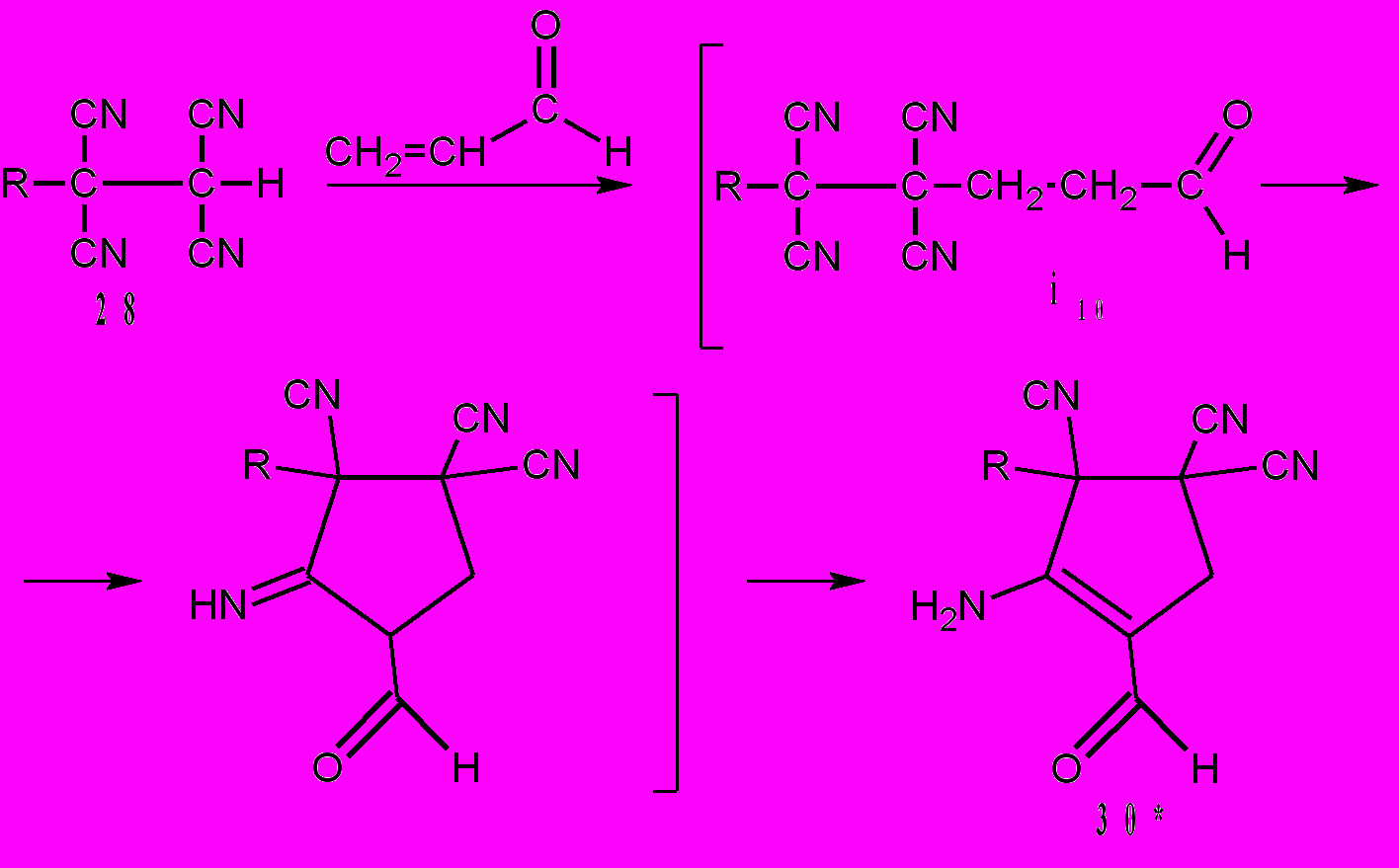
О преимущественном взаимодействии гидроксила с цианогруппой в положении 3 интермедиата i9 (R = R1 = R2 = CH3) свидетельствуют компьютерные расчеты межатомных расстояний. Их наименьшие значения между ОН и CN в положениях 1,3 равны – 2,82 Å, ОН и CN в положениях 1,4 равны – 4,49Å. Эти расчеты находятся в соответствии с рентгеноструктурными исследованиями соединения 29н. Проводя дальнейшие исследования свойств тетрацианоалканонов, мы разработали новый метод трансформирования кетонов, в том числе природных и биологически активных, в производные 4-формил-3-циклопентен-1,1,2-трикарбонитрилов. Реакция алканонов 28 с акролеином приводит к циклопентенам 30. Полагаем, что вначале образуется аддукт Михаэля i10. Затем в результате процессов циклизации и изомеризации образуются циклопентены 30. Эта реакция специфична тем, что двойные связи С=С и С=О в алканонах 28 не затрагиваются. По данным ТСХ соединения 28 превращаются в циклопентены 30 при комнатной температуре за 20-30 мин.
Схема 13.
| 30: | 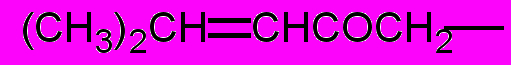 | (а)*; | R= | 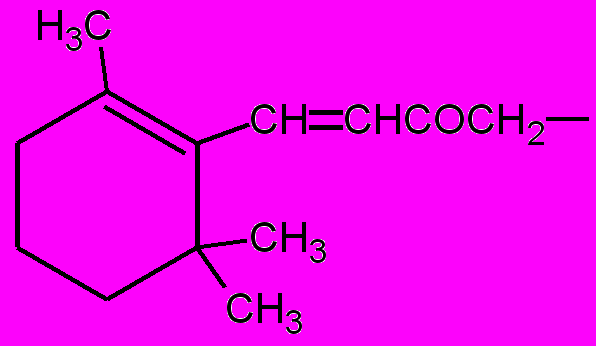 | (б); |
| R = | |||||
| R = | 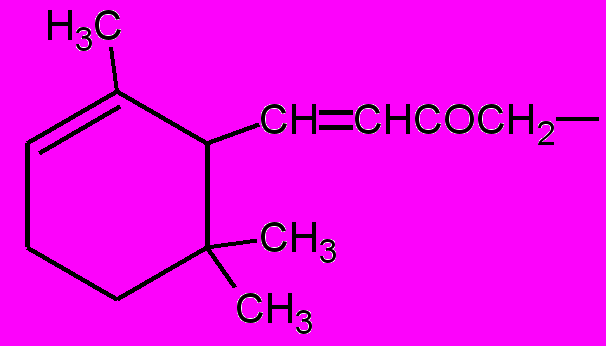 | (в); | R = | 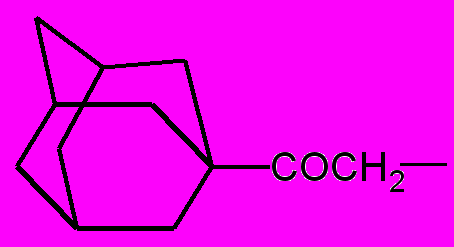 | (г); |
| R = |  | (д); | R = |  | (е)*. |