
Патогенетические обоснования стратегии и тактики лечения миастении 14. 00. 16 патологическая физиология 14. 00. 13 нервные болезни
| Вид материала | Автореферат диссертации |
- Ов курса «Патологическая физиология» для самостоятельного изучения, 13.37kb.
- Клинико-патогенетическое обоснование комбинированного лазерно-медикаментозного лечения, 270.48kb.
- Пояснительная записка к учебной дисциплине Эпизоотология и инфекционные болезни животных, 166.41kb.
- Пояснительная записка Учебно-методический комплекс по курсу «Патологическая физиология», 140.7kb.
- «Патогенетические механизмы развития хронической обструктивной болезни лёгких», 323.59kb.
- Клинико-патогенетические механизмы развития внешне и внутрисекреторной недостаточности, 559.83kb.
- Методические рекомендации для практического занятия №17, №18 Тема: Методика обследования, 25.33kb.
- Задачи по оказанию доврачебной помощи при неотложных состояниях 43 акушерство, 1884.21kb.
- Патогенетические механизмы развития микроциркуляторных нарушений в нефроне и периферических, 264.59kb.
- Патогенетическое обоснование восстановительного лечения нефропатий, сочетанных с нарушениями, 773.4kb.
Публикации: по материалам диссертации опубликовано 32 работы.
Объем и структура диссертации Диссертация состоит из введения, обзора литературы, главы с изложением материалов и методов, 7 глав собственных исследований, обсуждения, выводов и списка литературы. Диссертация изложена на 326 страницах машинописного текста, иллюстрирована 67 рисунками и 19 таблицами. Библиография включает в себя 501 источник литературы, из них 56 – отечественных и 445 – зарубежных авторов.
МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Обследование пациентов и контрольных лиц проводилось в Центре нервно-мышечной патологии НИИ общей патологии и патофизиологии РАМН и неврологических отделениях ЦКБ МПС России.
Группу исследования составили 422 больных миастенией в возрасте от 2,5 до 80 лет (средний возраст, 37,1 ± 20,5 лет, M ), наблюдавшихся во Всероссийском Миастеническом центре НИИ ОП и ПФ РАМН с 1997 по 2007 гг. Количество женщин преобладало над мужчинами в 3 раза. Приобретенная аутоиммунная миастения наблюдалась у 401 пациента, врожденные миастенические синдромы в 21 случаях. Тимома обнаруживалась у 57 больных (14,2%).
Электромиографически использовался метод непрямой ритмической супрамаксимальной стимуляции мышцы с регистрацией М-ответа поверхностными электродами (декремент-тест). Ритмическая стимуляция мышц проводилась на отечественном приборе “Нейромиан - МЕДИКОМ” производства России (Таганрог). Обследовано 1082 мышцы. Электромиографическое обследование проводилось в клинически пораженных мышцах у каждого пациента. При выраженной мимической слабости и «гнусавости» исследовались мышцы, иннервируемые лицевым нервом (m.orbicularis oculi, m.orbicularis oris, m. nasalis); при нарушениях функции мышц, удерживающих нижнюю челюсть, и жалобах на затруднения жевания - m. digastricus (n.trigemenus); при слабости мышц шеи – m. trapezius (n.accessorius), при выраженной слабости в проксимальных отделах рук - m. deltoideus (n.axillaris), m.triceps brachii и m.anconeus (n.radialis), при снижении силы в кистях рук - m. abductor digiti minimi (n.ulnaris).
Сила мышцы определялась по шестибалльной шкале. Изучение функционального состояния нервно-мышечной передачи оценивалось по следующим параметрам: амплитуде негативной фазы М-ответа в ответ на одиночный супрамаксимальный стимул (мВ); величине декремента амплитуды М-ответа при стимуляции мышцы частотой 3 имп/с по отношению 5-го М-ответа к 1-му (в %); изменению амплитуды М-ответа при стимуляции частотой 3 имп/с через 2 с после окончания тетанической серии или максимального произвольного усилия (в течение 10 с) (посттетаническое или постактивационное облегчение) по отношению к исходному М-ответу и 5-го М-ответа к 1-му (в абсолютных величинах и в %); изменению амплитуды М-ответа в серии из 5 импульсов при стимуляции частотой 3 имп/с через 3 мин. после окончания тетанической серии или максимального произвольного усилия (посттетаническое или постактивационное истощение) - по отношению к исходному М-ответу и 5-го М-ответа к 1-му (в абсолютных величинах и в %); изменению амплитуды М-ответа и декремента амплитуды М-ответа после подкожного введения адекватной дозы прозерина по отношению к исходному М-ответу и 5-го М-ответа к 1-му (в абсолютных величинах и в %).
Для объективизации нарушений дыхания у больных миастенией была разработана методика изучения нервно-мышечной передачи в диафрагме с использованием данных, взятых из литературных источников (Mier A., et al., 1992; Zifko U., et al., 1999), рис. 1.
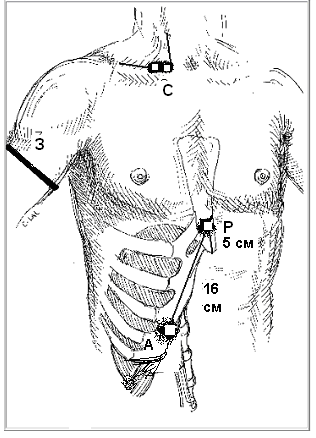
Рис.1. Схема наложения электродов при исследовании нервно-мышечного проведения «диафрагмальный нерв – диафрагмальная мышца»
Р – место расположения референтного электрода (накладывается на 5см выше нижнего края мечевидного отростка), A – место расположения активного электрода (реберный край, на расстоянии 16 см от референтного электрода). С – область расположения стимулирующего электрода (проекция пересечения латерального края грудино-ключично-сосцевидной мышцы и ключицы, в стороне от плечевого сплетения (на 2-4 см медиальнее от точки Эрба); З – место расположения заземляющего электрода (проксимальные отделы плеча).
Ритмическая стимуляция диафрагмального нерва сериями импульсов низкой частоты (3 имп/с) выполнялась на одной стороне. Патологией считалась величина декремента амплитуды и площади М-ответа более –11% (Zifko U., et al., 1999), рис.2.
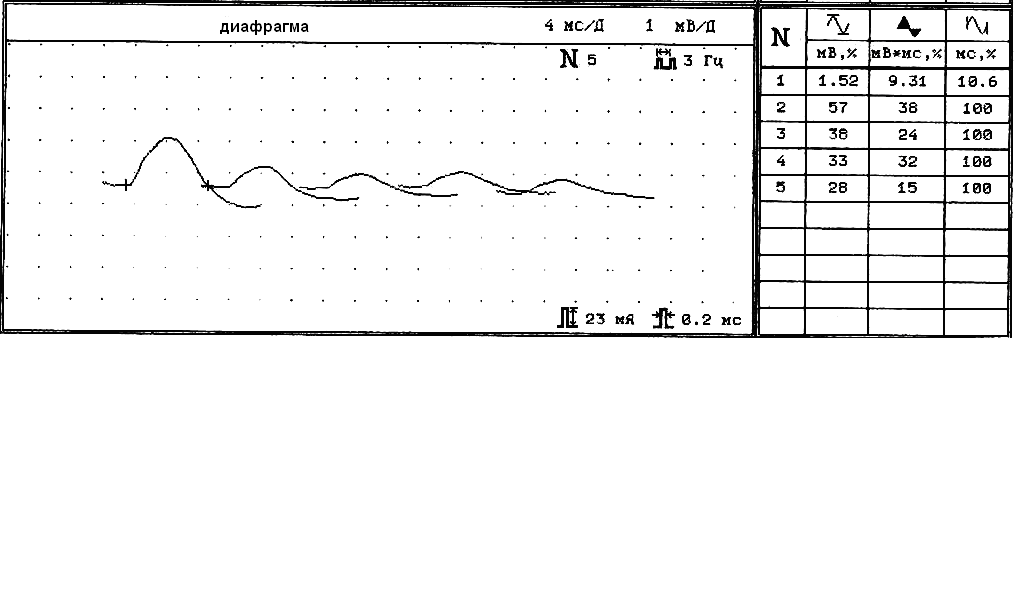
Рис.2. Изменение М-ответа диафрагмальной мышце при стимуляции частотой 3 имп/c у больной К., 26 лет.
Амплитуда первого М-ответа 1,52 мВ, площадь 9,31 мвмс, длительность 10,6 мс, декремент М ответа по амплитуде –72%, по площади -85%, по длительности 0%.
Для изучения феномена «медленных каналов» ацетилхолиновых рецепторов у больных с врожденной и аутоиммунной миастенией с использованием материалов, взятых из литературных источников (Kimura J. et al., 1978; Kimura J., 1981; van Dijk J.G. et al., 1994; 1995; 1996; 1997; 1999; Bromberg M.B., Spiegelberg T., 1997; Amoiridis G., Vlachonikolis I.G., 2003), была разработана техника регистрации повторных М-ответов, рис.3.
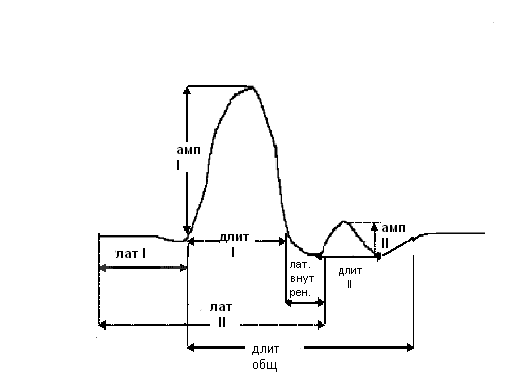
Рис. 3. Схема измерения основных параметров повторных М-ответов
ЛатI и Лат II (латентность1-го и 2-го ответов) промежуток времени между подачей стимула и возникновением I и II М-ответа, соответственно, в мсек.; Лат. внутр.(внутренняя латентность) - промежуток времени между окончанием основного (I) и началом дополнительного (II) М-ответов; амп I и длитI – амплитуда и длительность негативной фазы I М-ответа при одиночной супрамаксимальной стимуляции, в мВ и мс; амп II и длит II – амплитуда и длительность негативной фазы II М-ответа при одиночной супрамаксимальной стимуляции, в мВ и мс.
Выявлены критерии достоверности повторного М-ответа:
- Достоверность повторного М-ответа признавалась, если, несмотря на проведение всех мер по его устранению (поиск двигательной точки с изменением положения активного электрода, исключения наложения электродов на анастомозы нервов, в частности “n.medians - n.ulnaris” анастомоз Мартина-Грубера, и в зоны перекрещивающейся иннервации соседних мышц), дополнительный компонент М-ответа продолжал воспроизводиться.
- Длительность латентности (внутренней латентности, рис.3) между основным и повторным компонентом М-ответа должна превышать абсолютный рефрактерный период мышечных волокон – 2,5-3,0 мс.
- Повторный компонент должен быть декрементным при ритмической стимуляции. Декремент повторного компонента должен преобладать над декрементом основного компонента М-ответа.
- Повторный компонент М-ответа должен увеличиваться при стимуляции стимулами с коротким межимпульсным интервалом (спаренные стимулы, 25мс), высокой частоте стимуляции (40 имп/c) в период ПАО (ПТО), а также на фоне введения прозерина.
Изучение функционального состояния потенциалов двигательных единиц мышечных волокон с использованием игольчатых электродов проводилось в лаборатории клинической электромиографии (руководитель – доктор биологических наук, профессор Л.Ф. Касаткина), на электромиографах Counterpoint фирмы Dantec Medical (Дания). Обследовано 148 мышц 72 больных миастенией.
Иммунологические обследования выполнялись в лаборатории полисистемных методов исследований (руководитель – д.б.н. М.Ю. Карганов) и в лаборатории нейроиммунопатологии (руководитель – д.м.н., профессор В.А. Евсеев). Концентрацию антител к ацетилхолиновому рецептору и мышечной тирозинкиназе определяли радиоиммунологическим методом, а уровень антител к мышечному белку титину - методом твердофазного иммуноферментного анализа. Иммунологическое обследование прошли 152 больных миастенией.
Молекулярно-генетический анализ прошли четверо больных из двух семей с клиническим диагнозом «врожденный миастенический синдром». Анализ проводился в генетическом центре Мюнхенского университета им. Людвига Максимилиана профессором Хансом Лохмюллером под руководством д.м.н. О.П. Сидоровой (МОНИКИ, Москва), Lochmuller H., Abicht A. (Мюнхен). Медико-генетический анализ был проведен с использованием геномной ДНК больных. У двух больных дефект нервно-мышечной передачи был вызван новой мутацией в сайте сплайсинга IVS7-2A/G гена ε-субъединицы АХР и мутацией ε1293inG со сдвигом рамки считывания генетической информации, описанной ранее (Sieb J. et al., 2000). У двух больных из второй семьи была определена уже известная гомозиготная точковая мутация в промоторном регионе гена -субъединицы АХР (Щербакова Н.И., и др.2004; Ohno K., et al., 1999).
Клинико-электрофизиологическое обследование. Все больные перед началом патогенетической терапии были обследованы до- и после введения прозерина. Проба с введением прозерина позволяла оценить обратимость и степень возможной компенсации синаптической функции. Введение прозерина проводилось на фоне отмены других антихолинэстеразных препаратов на максимально длительный срок. Препарат вводился подкожно в дозе 0.125 мг на 1кг массы тела (1.5-2.0 мл 0.05% раствора). Электромиографическое тестирование состояния нервно-мышечной передачи проводилось до введения прозерина и через 40 минут после введения препарата.
285 больных (71,1%) прошли курс лечения преднизолоном (метилпреднизолоном) - прием per os в дозе 1 мг/ 1 кг массы тела. Больные обследованы клинико-электромиографически через 10 и 30-60 приемов преднизолона (метилпреднизолона).
Тим(тимом)эктомия проведена 156 пациентам (36,1%). Клинико-электромиографическое обследование проводилось через 10-14 дней и спустя 6-12 месяцев после тим(тимом)эктомии.
Клиническое и электромиографическое исследование эффекта цитостатической терапии (азатиоприн, циклофосфан, сандиммун неорал) проводилось на разных сроках от начала лечения (от 1 месяца до 3-х лет).
Лечение азатиоприном (имуран) прошли 57 (14,1%) больных миастенией. Суточная доза варьировала от 1 до 5 мг/кг массы тела. Продолжительность приема насчитывала от 2-х до 14 лет. Монотерапию азатиоприном проходили 6 больных, 51 получали препарат на фоне базисного приема преднизолона (метипреда).
Парентеральное лечение циклофосфаном в дозе 200 мг внутримышечно ежедневно проводилось 53 (13,1%) пациентам. Монотерапию циклофосфаном проходили 9 больных, 44 получали препарат на фоне базисного приема преднизолона (метипреда). Продолжительность лечения препаратом составляла от 2-х недель до 3-х лет, суммарная доза – от 800 мг до 30г 600мг.
Курс лечения циклоспорином А (сандиммун неорал) прошли 31 (7,7%) больных. Суточная доза 2,5-5 мг/кг массы тела. Продолжительность приема сандиммуна у разных пациентов колебалась от 4 месяцев до 7 лет.
Внутривенное введение нормального человеческого иммуноглобулина G (октагам, вигам, биавен) проводилось 25 больным (6,2%). Количество вводимого препарата варьировало от высоких доз (400 мг на 1кг массы тела в течение 5 дней) в виде монотерапии 5 пациентам - до минимальных (0,4-0,5 мг на 1кг через 1-2 дня в течение 7-18 дней) на фоне базисного приема глюкокортикоидных стероидов (преднизолон, метилпреднизолон) 20 пациентам. Объем однократного внутривенного введения варьировал от 1 до 24г, суммарная доза варьировала от 12,5 до 120г.
Все полученные результаты были обработаны статистически на ПЭВМ IBM PC/AT с помощью статистических программ (CSS и Stat). Вычислялись средние величины и их разброс (М сигма). Изучалась также корреляция между различными показателями (с вычислением коэффициента корреляции - “r” и степени его достоверности - “p” с применением t-критерия Стъюдента). Достоверными считались изменения при p<0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ И ИХ ОБСУЖДЕНИЕ
Современные представления о неоднородности патофизиологических механизмов миастении (Санадзе А.Г. и др., 2003; 2006; Vincent A. et al., 2006; Conti-Fine B.M., Kaminski H.J., 2006; Farrugia M.E. et al., 2007), неоднозначность эффекта основных видов лечения на различных больных - от полного выздоровления у одних пациентов, небольшого улучшения или отсутствия эффекта у других, до развития обострений заболевания с миастеническими кризами у третьих (Санадзе А.Г. и др., 2005; Mann J.D., et al., 1976; Johns T.R., 1987; Schneider-Gold C. et al., 2005; Punga R.A. et al., 2006), указывают на необходимость поиска дополнительных патогенетических обоснований стратегии и тактики лечения заболевания, требует уточнения критериев целесообразности проведения таких важных мероприятий как тимэктомия, внутривенное введение высоких доз глюкокортикоидных стероидов, оправданности назначения дорогостоящих цитостатических иммуносупрессантов и человеческих иммуноглобулинов.
Разнообразие антигенных мишеней при миастении предполагает наличие различных вариантов патологии синапса, определяя актуальность поиска патофизиологических механизмов нарушений нервно-мышечного проведения, позволяющих целенаправленно регулировать эффективность синаптической передачи.
Совокупность результатов электрофизиологических, иммунологических и клинических исследований 422 больных генерализованной миастенией позволило выявить три типа патологии синапса.
I тип – классический постсинаптический тип патологии синапса, описанный большинством исследователей (Гехт Б.М., 1990, Санадзе А.Г., 2001; Krarup C., 1984; Maselli R.A., 1998; Oh S.J. et al., 2006), доминировал у исследуемых больных миастенией (275 больных, 65% случаев). Основной клинико-электрофизиологической характеристикой I типа была высоко достоверная корреляция между степенью снижения силы мышцы и величиной декремента М-ответа при стимуляции 3 имп/c (r = -0,73, P<0.001). Средние значения амплитуды и площади М-ответа на одиночный супрамаксимальный стимул соответствовали физиологической норме для каждой из исследуемых групп мышц. Прирост амплитуды и площади М-ответа в период постактивационного (посттетанического) облегчения (ПАО (ПТО)) не превышал 150%, в среднем составляя 115,5±15,7%. Введение прозерина приводило к различной степени восстановления силы мышц у всех больных с I типом патологии синапса, что было сопоставимо с уменьшением блока нервно-мышечной передачи. Изучение характера организации потенциалов двигательных единиц (ПДЕ) мышечных волокон в клинически пораженных мышцах у больных с первым типом поражения синапса показало отсутствие изменений показателей длительности и амплитуды ПДЕ, не выявило увеличения количества полифазных, псевдополифазных потенциалов и спонтанной активности мышечных волокон, что соответствовало физиологическим параметрам нормы для исследованных мышц. Клинико-электрофизиологическое сопоставление показало, что наиболее поражаемая мышца у больных миастенией при I типе патологии синапса является m.triceps brachii (n.radialis).
Исследование титра АТ к АХР в сыворотке больных с I типом выявило его увеличение в 100% случаях, при этом средние значения составили 7,97± 4,20 Нмоль/л, минимальное 0,60 Нмоль/л, максимальное 18,47 Нмоль/л (при норме < 0,5 Нмоль/л). Повышение уровня АТ к мышечному белку титину отмечалось в 37% случаях и составляло в среднем 1,20 ± 0,64 УЕ, минимальный уровень 0,26 УЕ, максимальный 3.9 УЕ (при норме < 1.0 УЕ).
Средний возраст начала заболевания с I типом патологии синапса составлял 36,54 ± 19,02 лет, отмечалось классическое преобладание женщин над мужчинами 3,3:1. Сочетание миастении с тимомой среди больных с I типом патологии синапса выявлялось в 2,5% случая (7 больных).
Клинически I тип патологии синапса характеризовался классическим распределением мышечной слабости с преимущественным поражением наиболее типичных для миастении мышечных групп: эстраокулярных в 62,9% случаях, проксимальных мышц рук и ног у 67,8%; бульбарных в 29,5%; наиболее частым дебютом заболевания с птоза и двоения, встречавшихся в 61 и 58% случаев, соответственно.
Таким образом, I тип олицетворяет собой строго постсинаптическую «модель» поражения синапса, при которой ключевую роль в снижении надежности нервно-мышечной передачи играет уменьшение числа функционирующих рецепторов АХ на постсинаптической мембране, электрофизиологическим эквивалентом которого является декремент М-ответа при стимуляции 3 имп/c. Обнаружение в сыворотке всех больных с I типом аутоантител, связывающих комплемент на постсинаптической мембране, указывало, что ведущим механизмом, приводящим к дефициту функционирующих АХР, является аутоиммунный процесс комплемент-опосредованного лизиса постсинаптической мембраны. Обнаружение у 37% больных АТ к титину было обусловлено наличием соответствующего количества больных с «поздним» началом заболевания, что указывало на клиническое многообразие I типа патологии синапса.
По уровню поражения синапса, большинство авторов (Гехт Б.М. и др., 2003; Lindstrom J.M., 2000; Vincent A. et al., 2000; 2006; Conti-Fine B.M., Kaminski H.J., 2006) относят миастению к исключительно постсинаптическому заболеванию, для которого не характерны нарушения пресинаптических механизмов выделения медиатора. Тем не менее, время от времени, в литературе появляются сообщения об обнаружении случаев неожиданного сходства электрофизиологического паттерна миастении с пресинаптическим синдромом Ламберта Итона (Singer P. et al., 1987; Санадзе А.Г., 1991; 2001; Matsumoto R. et al.,1999; Sha S.J., Layzer R.B., 2007).
В нашем исследовании из всей совокупности больных выделялась группа (22% пациента), характер поражения нервно-мышечной передачи, которых указывал на дополнительное вовлечение пресинаптических механизмов поражения синапса.
II тип патологии синапса (99 больных, 22% случаев) имел несколько важных отличий. Так, классическая корреляция между степенью снижения силы мышцы и величиной декремента М-ответа при стимуляции 3 имп/c не наблюдалась (P>0,05). При этом выраженность декремента, как правило, преобладала над снижением силы мышцы. Больные могли не жаловаться на слабость в мышце и объективного снижения силы в мышце не обнаруживалось, при этом величина блока нервно-мышечной передачи достигала порядка -50%. Так, если у пациентов с I типом патологии синапса, средние значения декремента М-ответа при отсутствии слабости в мышце (сила 5 баллов) составляли - 5,5± 4,7%, то у пациентов со II типом при той же силе декремент составлял -48,7±8,3 (t= 4.53, р<0,001). Это указывало на то обстоятельство, что декремент М-ответа не являлся объективным критерием патологического процесса в синапсе при II типе поражения, и позволяло предполагать о существовании дополнительных компенсаторных механизмов функционирования синапса, «маскирующих» слабость мышцы. Обращало внимание снижение средних значений амплитуды и площади М-ответа на одиночный супрамаксимальный стимул у больных со II типом патологии синапса, по сравнению с I типом. Так, у больных с I типом поражения синапса в мышцах, имеющих одинаковые нижние границы нормы -4,5-5,0мВ (m. deltoideus, m.triceps brachii, m. abductor digity minimi), средние значения амплитуды М-ответа при силе 5 баллов составляли 12,8±2,7 мВ. При этом в тех же мышцах и при той же силе у больных со II типом средняя величина М-ответа была снижена - 3,8±1,6 (t=2.90, P<0,05). Наиболее важные отличия были обнаружены в условиях, облегчающих выделение медиатора, после максимального мышечного усилия (ПАО) или тетанической стимуляции (150-200 стимулов с частотой 40 имп/c) (ПТО). По сравнению с физиологическим приростом амплитуды М-ответа в период ПАО (ПТО) - 115,5±15,7% у больных с I типом патологии синапса, пациенты со II типом имели патологическое увеличение феномена фасилитации (средние значения, 329,1±73,9%), что достоверно (t=2.92, P<0,05) отличало характер нарушений нервно-мышечной передачи пациентов II типа от I. Столь значительный прирост М-ответа в период облегчения выделения медиатора, по-видимому, «маскировал» слабость в мышце при клиническом измерении силы, которое воспроизводит феномен максимального мышечного усилия (ПАО).
Характер нарушений нервно-мышечной передачи у больных со II типом патологии синапса обнаруживал сходство с пресинаптическим миастеническим синдромом Ламберта Итона (МСЛИ), однако, в отличие от последнего, выявлялся не во всех мышцах у одного и того же пациента. Так, по нашим данным чаще всего патологически увеличенный феномен ПАО можно было обнаружить в m. deltoideus (n.axillaris), мышце, наиболее вовлеченной в патологический процесс у пациентов со II типом поражения (t=3.1, P<0,01). У больных со II типом не отмечалось характерного для МСЛИ снижения или выпадения сухожильных рефлексов. Кроме того, исследование титра АТ к АХР в сыворотке больных с II типом выявило увеличение концентрации АТ к АХР в 95% случаях, при этом средние значения составили 14,37± 6,20 Нмоль/л, минимальное 4,0 Нмоль/л, максимальное 29,87 Нмоль/л, (при норме < 0,5 Нмоль/л), что подтверждало диагноз миастении. Таким образом, II тип патологии синапса характеризовался наличием у одного и того же больного мышц с сочетанной пост - и пресинаптической патологией нервно-мышечного проведения и мышц, с классическим постсинаптическим характером поражения. Введение прозерина приводило к неравномерному восстановлению мышечной силы и обратимости нарушений нервно-мышечной передачи: от полной компенсации в мышцах с постсинаптическим характером поражения, до - частичной – в мышцах с сочетанной патологией.
Изучение характера организации ПДЕ мышечных волокон в дельтовидной мышце у больных со II типом патологии синапса выявило признаки вовлечения в патологический процесс мышечного субстрата: снижение средней длительности ПДЕ на 18,3 7,6%, увеличение процента ПДЕ с минимальной средней длительностью (51,8 22,2% от всех потенциалов); некоторое увеличение полифазных 16,410,2% и псевдополифазных потенциалов 20,715,1%, появление единичной спонтанной активности мышечных волокон в виде потенциалов фибрилляций (ПФ) и положительных острых волн (ПОВ). Кроме того, выявлялась популяция ПДЕ с минимальной средней длительностью, которая не восстанавливалась после введения антихолинэстеразных препаратов. Повышение уровня АТ к мышечному белку титину, которое отмечалось в 70% случаях и составляло в среднем 2,08 ± 0,94 УЕ, (минимальный уровень 0,7 УЕ, максимальный 4.5 УЕ), подтверждало аутоиммунные механизмы вовлечения мышечного компонента в патологический процесс у больных со II типом патологии синапса.
Обращало внимание преобладание среди пациентов со II типом типичного для миопатии преимущественно туловищного распределения мышечной слабости у 65,4% больных, а в 11% случаев выявлялись чисто туловищные формы, описанные в литературе как «конечностно-поясные» ("limb-girdle" form ) формы миастении c ювенильным началом заболевания (Oh S.J., Kuruoglu R., 1992; Rodolico C. et al., 2005; Slater C.R. et al., 2006). II тип выявлялся в 100% случаях аутоиммунных семейных форм заболевания (8,7% от всех больных со II типом).
Периодически скрытые туловищные нарушения обнаруживались у больных с клиническими проявлениями локальной глазной и «глазо-дистальной» (Перельман Л.Б. и др., 1979; Гехт Б.М. и др., 1983) формами миастении (5,5% и 12,3% случаев соответственно), когда неожиданно выявлялись электрофизиологические признаки сочетанной патологии рецепции и секреции медиатора в клинически интактных проксимальных мышцах конечностей.
Миастения при II типе наиболее часто дебютировала со слабости в проксимальных отделах ног (61,7%) и рук (41%), слабости мышц шеи (31%). Из-за нетипичного распределения мышечной слабости, имитирующего миопатию, диагноз миастении ставился с задержкой от 9 месяцев до 5 лет. Начало заболевания с двоения и птоза, типичное для I типа патологии синапса, у больных со II типом встречалось значительно реже, в 33,3 и 22,0% соответственно. Средний возраст дебюта заболевания со II типом патологии синапса был старше (41,47 ± 29,02 лет), а преобладание женщин над мужчинами было менее выраженным - 2:1, по сравнению с I типом.
Сходство электрофизиологического профиля и клинического паттерна с пресинаптическим миастеническим синдромом Ламберта-Итона (МСЛИ) указывало на необходимость поиска патогенетической связи между миастенией со II типом поражения синапса и МСЛИ.
Клинический синдром, развивающийся при МСЛИ, обусловлен нарушением квантового выделения ацетилхолина из пресинаптических окончаний, вследствие аутоиммунной агрессии аутоантителами (Ig, класса G) P/Q и N – типов потенциалзависимых кальциевых каналов (ПЗКК), расположенных в терминалях аксонов (Санадзе А.Г. и др., 2006; Lennon V.A., 1995; Lang B. et al., 2003). При этом у 85% больных принципиальной мишенью для ауто-АТ служит a1A субъединица P/Q-типа, где и располагается основной канал, по которому кальций входит в терминаль аксона (Pinto A., Lang B., 2003). Несмотря на существующее разделение больных с МСЛИ на 2 подгруппы: 1) паранеопластический МСЛИ, ассоциируемый, как правило, с мелкоклеточной карциномой легких и 2) МСЛИ без неопластического процесса, - оба эти состояния имеют общую паранеопластическую природу (Lindstrom J., 1997; Vinsent А., 1999; Nishimura T. et al., 2006; Vernino S., 2007).
Изучение паранеопластического фона у больных исследуемой группы показало, что частота сочетания миастении с тимомами у пациентов со II типом патологии синапса составила 55,5±10,6%, что с высокой статистической достоверностью (t=4.8, P<0,001) преобладало над обнаружением подобных случаев среди больных с классическим I типом (2,5±3,1%). Сочетание миастении с «нетимическими» неоплазиями также достоверно (t=3.98, P<0,001) преобладало у пациентов со II типом патологии синапса, составляя 28,2±5,2% случаев, по сравнению с 4,3 ± 3,1% у больных с I типом патологии синапса.
Таким образом, электрофизиологическое сходство II типа патологии синапса с МСЛИ было не случайным и указывало на общую паранеопластическую природу патологии синапса (рис.4,5).
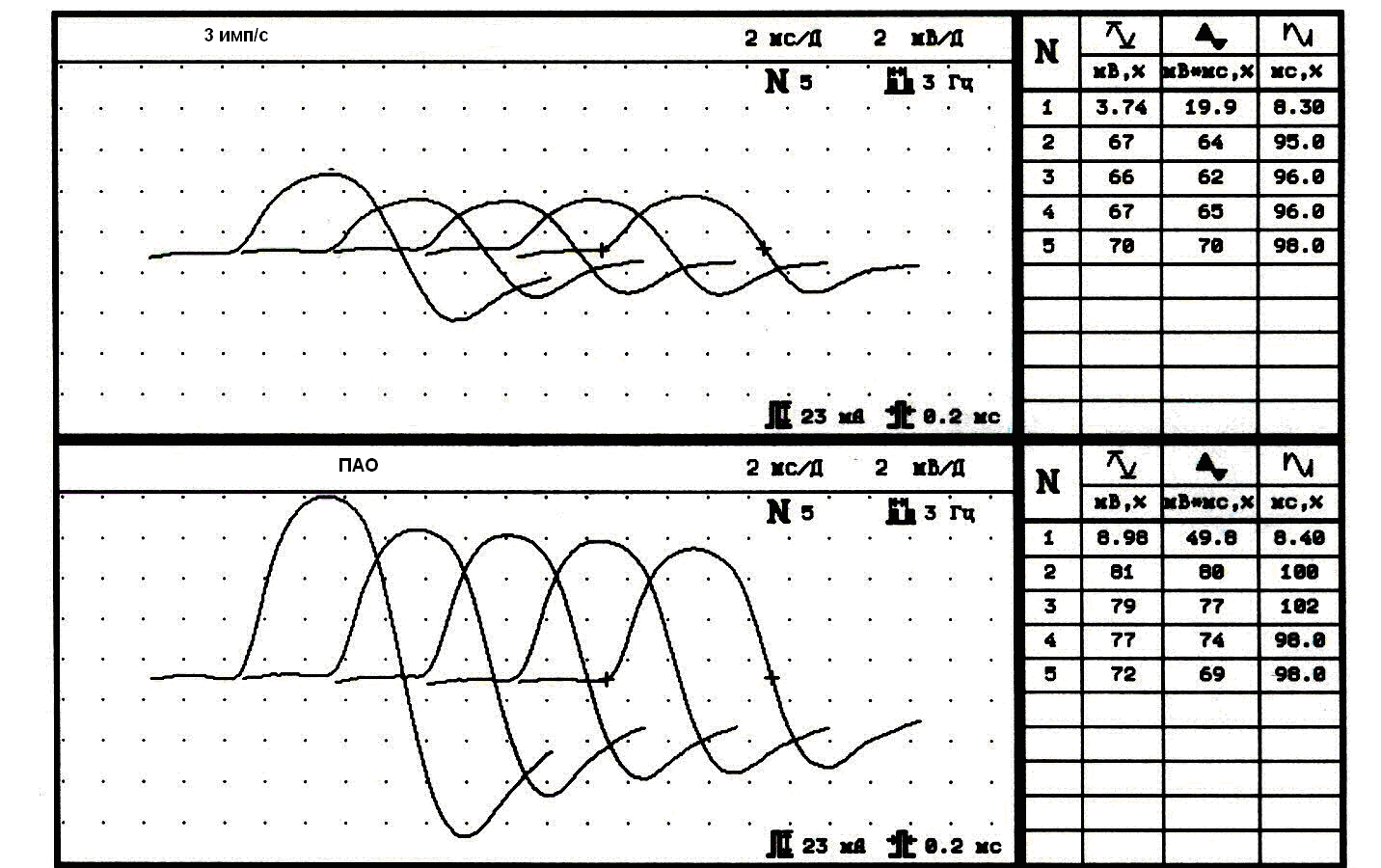
Рис 4. Увеличение М-ответа на 240% в период ПАО в дельтовидной мышце у пациентки К., 50 лет, имеющей сочетание миастении с тимомой.
В верхней части рисунка изменение М-ответа дельтовидной мышцы при стимуляции 3 имп/с (амлитуда 3,74 мВ, площадь 19,9 мвмс 1-го М-ответа, декремент 5-го М-ответа -30%). В нижней части рисунка - феномен ПАО: увеличение амплитуды до 8,98 мВ (240%) и площади до 49.8 мвмс (250%).
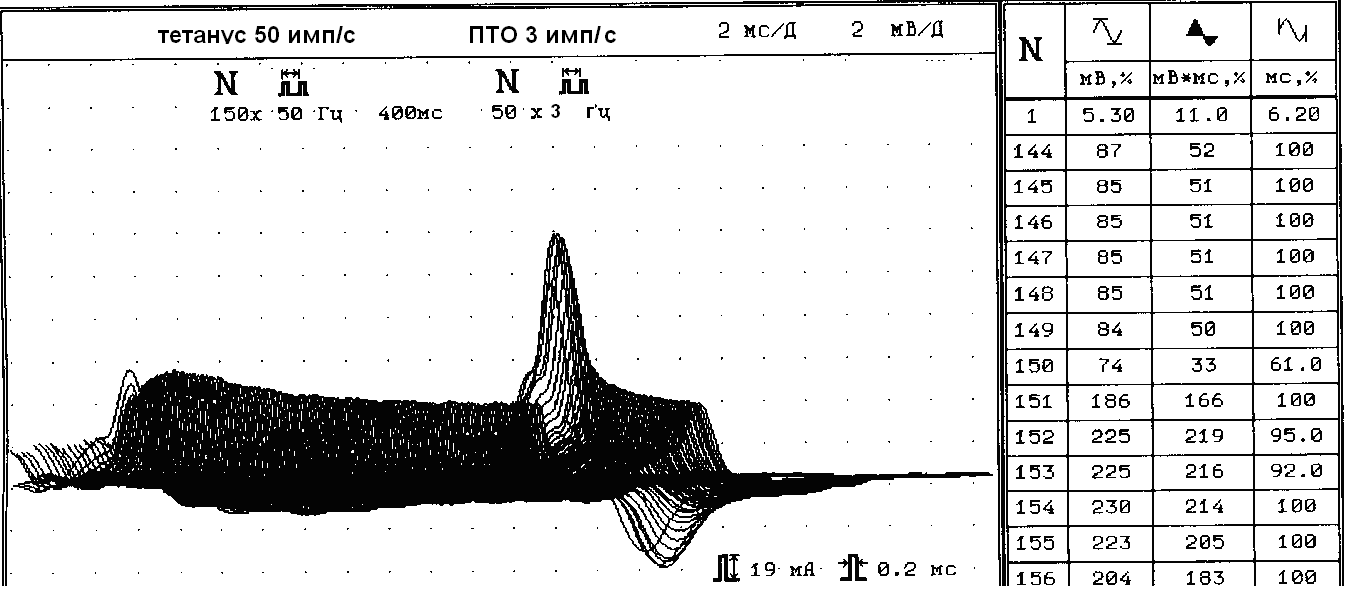
Рис. 5. Увеличение М-ответа на 225% в период ПТО в мышце, отводящей мизинец кисти у больной М., 42 лет с сочетанием миастении и рабдомиосаркомы бедра.
Слева направо показано изменение М-ответа при стимуляции 150 стимулами частотой 50 Гц (тетанус) с дальнейшей стимуляцией 50 стимулами с частотой 3 имп/сек. В период тетануса амплитуда М-ответа колебалась в пределах 5,3 мВ ± 25%; в период посттетанического облегчения (ПТО) выявляется феномен патологически увеличенного ПТО: увеличение амплитуды 152-го М-ответа на 225% по амплитуде и на 216% по площади с характерным для синдрома Ламберта-Итона декрементом в период ПТО (феномен «паруса»).
При этом из 40 случаев экстратимических новообразований («cancer» - cr) 28 (70%) опережало начало миастении на 2-12 лет: cr матки и яичников (33,3%), cr молочной железы (7,7%); cr простаты (5,1%); cr почек (7,7%); cr желудка (7,7%), cr щитовидной железы (7,7%), cr легкого (7,7%). У 12 других больных (30%) начало миастении практически совпадало или несколько опережало (от 4-х месяцев до 1,5 года) обнаружение онкологических заболеваний: базалиомы кожи в 3 случаях (7,7%); гемангиомы спинного мозга 3 случая (7,7%); лимфогрануломатоза 2 случая (5,1%), B-клеточных лимфом переднего средостения и головного мозга (5,1%), рабдомиосаркомы бедра в 1 случае; аденокарциномы сигмовидной кишки в 1 случае.
У трех больных, имеющих наибольшее электрофизиологическое сходство с МСЛИ, миастения которых в одном случае развивалась на фоне базалиомы кожи лица; в другом - рабдомиосаркомы бедра; в третьем -лимфоэпителиальной тимомы, - была исследована сыворотка крови на наличие АТ к АХР, титину и кальциевым каналам P/Q типа. Титр АТ к АХР и уровень АТ к титину оказались повышенными (средние значения 9,27± 8,2 Нмоль/л и 2,07± 0,5 УЕ, соответственно). При этом АТ к кальциевым каналам P/Q типа не выявлялись ни в одном случае, что подтверждало диагноз миастении, а не МСЛИ.
Обнаружение у больных миастенией клинических и электрофизиологических проявлений МСЛИ в литературе описано крайне редко. При этом в основном встречаются примеры, когда миастения через какое-то время трансформируется в МСЛИ (Санадзе А.Г., 1991; Newsom-Davis J. et al., 1991; Matsumoto R. et al., 1999). Тем не менее, одновременное сосуществование у больных миастенией пост - и пресинаптических нарушений показано в ряде работ (Emeryk В. et al., 1991; Oh S.J., Sher E., 2005; Sha S.J., Layzer R.B., 2007). При этом S.J. Oh и E. Sher (2005), J. Newsom-Davis c коллегами (1991) приводят иммунологические доказательства сосуществования миастении и МСЛИ, выявляя одновременно АТ к АХР и N-типу кальциевых каналов. Подтверждения неопластической этиологии миастении с чертами МСЛИ мы нашли в исследованиях J.P. Ferroir и соавт., (1999), J.A. Leavitt (2000) и F. Roohi и соавт., (2006). Leavitt J.A. (2000) описывает случай развития подобного сочетанного синаптического дефекта, развившегося спустя 3 года после удаления крупноклеточной карциномы легких. F. Roohi с коллегами (2006), докладывают о параллельном развитии миастении с чертами МСЛИ на фоне лейомиосаркомы матки, а J.P. Ferroir с коллегами (1999) - на фоне нейроэндокринной мелкоклеточной карциномы легких. Во всех этих случаях, наличие АТ к АХР и негативный тест на АТ к кальциевым каналам P/Q типа подтверждало диагноз миастении, а не МСЛИ.
Таким образом, столь категоричное разделение нервно-мышечных заболеваний на постсинаптические и пресинаптические не всегда правомочно, в ряде случаев опровергается сосуществованием пре- и постсинаптического дефекта нервно-мышечной передачи, у одного и того же пациента и требует повышенного внимания в связи с высокой вероятностью паранеопластического фона.
Между тем, нарушение выделения медиатора может иметь и не иммунную природу, а являться следствием патологии процессов ауторегуляции секреции АХ в синапсе, обусловленных изменением метаболизма синаптического аппарата у больных с неоплазиями. Важную роль играет энергетическое обеспечение секреторного аппарата, которое, вполне вероятно, изменяется на фоне онкологических процессов. Синтез АХ существенно зависит от наличия глюкозы и состояния цитохромной системы (Крыжановский Г.Н. и др., 1974; Gage P.W., Hubbard J.I., 1966) и значительно снижается при нарушении окислительного фосфорилирования, которое сопровождается быстрым уменьшением содержания АТФ в терминалях аксона (Каменская М.А., 1972; Beani L. et. al.,1966). АТФ играет важную роль в освобождении медиатора. E.M. Silinsky, R.S. Redman, (1996) показали, что вместе с АХ двигательные нервные окончания выделяют также АТФ, который действует в качестве второго нейромедиатора, модулируя выброс АХ, вызывая пресинаптическое торможение. АТФ гидролизуется до аденозина, который затем обратно захватывается пресинаптической терминалью и уменьшает квантовый состав ответа. Кроме того, АТФ и сам может угнетать секрецию медиатора через собственные метаботропные пуриновые Р2 рецепторы (Giniatullin R.A. et al., 1997; 2001; 2005).
Таким образом, нарушения выделения медиатора при II типе патологии синапса могут быть обусловлены не только аутоиммунными процессами, но и поражением механизмов ауторегуляции медиатора на фоне изменения энергетического обеспечения секреторного аппарата в условиях паранеопластического процесса.
Постсинаптические механизмы поражения синапса у больных миастенией могут быть достаточно неоднородны. Еще в 1976г. D. Grob и T. Namba указывали на повышенную готовность АХР при этом заболевании к развитию десенситизации, предполагая о существовании популяций «патологических форм АХР» («abnormal forms of receptor») или АХР с патологически измененным ответом на медиатор. Несмотря на существующие представления о дефиците АХР на постсинаптической мембране, M.K. Palaga, T. Namba, D. Grob, (1981) обнаружили достаточное количество АХР и даже выявляли существования запасного (резервного) пула АХР, не участвующих при ритмической стимуляции. Эти исследователи впервые предположили о поражении не количества, а функции ионных каналов АХР у ряда больных миастенией - десенситизации АХР как ведущем механизме развития нервно-мышечного блока в отдельных группах заболевания.
В своем исследовании мы выделили группу больных, у которых при выраженной слабости в мышцах выявлялась очень незначительная степень декремента М-ответа при стимуляции 3 имп/c (средние значения -14,2±7,4%). Это позволяло предположить, что классический для миастении дефицит АХР на постсинаптической мембране, эквивалентом которого служит декремент М-ответа, не являлся ведущим патогенетическим механизмом поражения синапса в этой группе пациентов. Наше предположение подтвердило отсутствие АТ к АХР в сыворотке этих пациентов, основной патогенетической причины дефицита АХР на постсинаптической мембране. Так, мы выделили еще один тип патологии синапса, который наблюдался у больных с миастенией, не имеющих АТ к АХР (серонегативная миастения).
III тип патологии синапса встречался в 13% случаях (60 больных), характеризовался нормальными или несколько сниженными параметрами М-ответа на одиночной супрамаксимальный стимул (11,0±7,4, мВ), физиологическим приростом М-ответа в период ПТО (ПАО) (111±26,3%) и небольшими значениями декремента М-ответа при стимуляции 3 имп/с (средние значения -14,1±7,2%) даже при выраженном снижении силы мышцы, вплоть до пареза.
Детальный анализ позволил обнаружить в мышцах этого типа еще одну особенность: М-ответ в ряде случаев имел дополнительные компоненты, описанные в литературе как повторные М-ответы - «Repetitive compound muscle action potentials» (R-CMAPs) (Engel A.G. et al., 1982; 2000; Oosterhuis H.J. et al., 1987; van Dijk J.G. et al., 1996; Lorenzoni P.J. et al., 2006; Punga A.R. et al., 2006). Изучение характеристик М-ответа при разных частотах стимуляции, в период проведения функциональных проб и на фоне прозерина показало зависимость величины дополнительного компонента R-CMAP от концентрации доступного АХ в зоне взаимодействия с АХР. Так, R-CMAP увеличивался в период, характеризующийся облегчением выделения медиатора из пресинаптической терминали – при высокочастотной стимуляции (40 имп/c) и в период ПАО или ПТО, а также в условиях ингибирования ацетилхолинэстеразы; и уменьшался в период депрессии выделения медиатора, связанной с истощением количества квантов медиатора и уменьшением эффективности выброса (ПАИ, ПТИ).
Наиболее важные отличия III типа патологии синапса были выявлены при изучении обратимости нарушений нервно-мышечной передачи на фоне антихолинэстеразных препаратов (АХЭП), когда выявились чувствительные и не чувствительные к АХЭП препаратам мышцы. В чувствительных к прозерину мышцах сохранялась положительная клиническая и электрофизиологическая реакция. В «прозерин-негативных» - отмечалась негативная клиническая и электрофизиологическая реакция в виде фасцикуляций и миокимий, обращало внимание увеличение длительности М-ответа на фоне прозерина, появление R-CMAP.
Повторные М-ответы являются патогномоничным электрофизиологическим признаком поражения ионного канала АХР и наиболее часто описаны при врожденном миастеническом синдроме (ВМС) «медленного канала», получившем свое название вследствие замедленного периода спада токов концевых пластинок (ТКП). С помощью методики пэтч-клэмп (patch clamp), позволяющей регистрировать активность одиночных ионных каналов (Neher E., Sakman B., 1976) было обнаружено пролонгированное во времени пребывание ионного канала АХР в открытом состоянии. По данным A.G. Engel и соавт., (2000), феномен повторных М-ответов на одиночный электрический стимул возникает, если длительность пролонгированных ТКП превышает рефрактерный период потенциала действия мышечных волокон.
Необычайное сходство нейро-электрофизиологических проявлений поражения синапсов у ряда больных миастенией с ВМС «медленного канала» обнаруживают многие исследователи, а некоторые авторы даже выделяют «приобретенный синдром медленного канала», как отдельный вариант аутоиммунной миастении с пролонгированным временем открытия ионного канала АХР(Wintzen A.R. et al., 1998; Scola R.H. et al., 2000).
Доказательства того, что не дефицит АХР, а поражения кинетики ионных каналов АХР является ведущим механизмом патологии синапса при серонегативной миастении, приводят ряд авторов (Barrett-Jolley R. et al.,1994, Plested C.P. et al., 2002; Vincent A. et al., 2004; Padua L. et al., 2006). Исследователи обнаруживают у большинства серонегативных больных в плазме «не IgG фактор», (предположительно Ig M), который ингибирует ионный канал АХР, уменьшает время открытия АХР, нарушая ток ионов Na+ в канале. При этом ингибирующий эффект «не IgG фактора» в отношении АХР сопоставим с эффектом десенситизации, вызываемым 100 microM никотина.
В последние годы установлено, что основной патогенетической мишенью при серонегативной (СН) миастении является рецептор постсинаптической мембраны – мышечная тирозинкиназа (“muscle-specific kinase”, MuSK), который принимает участие в формировании кластеров АХР на постсинаптической мембране при участии агрина и рапсина (DeChiara T.M. et al., 1996; Glass D.J. et al., 1996). Антитела к MuSK обнаруживают у 40-70% больных СН миастенией. По современным представлениям эти АТ нарушают образования группировок (кластеров) АХР, когда АХР начинают распределяться диффузно вдоль всей поверхности миофибриллы, как в денервированной мышце (Sanders D.B. et. al., 2003; Shigemoto K. et al., 2006; Jha S. et al., 2006; Farrugia M.E. et al., 2007). Предполагается, что один из возможных эффекторных механизмов АТ к MuSK связан с повышением фосфорилирования АХР, что меняет конформационные свойства ионного канала АХР и повышает готовность к десенситизации (Li Z. et al., 1996; Farrugia M.E., Swingler R.J., 2002; Vincent A., et al., 2003). При этом по данным H. Shiraishi с коллегами (2005), А.Vincent, M.I. Leite, (2005), J. McConville с коллегами (2004), биопсия межреберных мышц и мышц конечностей СН пациентов демонстрирует отсутствие дефицита АХР. Авторы не находят депозитов комплемента на постсинаптической мембране и морфологических повреждений нервно-мышечного синапса, связывая это с тем, что анти-MuSK-АТ являются, главным образом, IgG4, которые не обладают способностью фиксировать комплемент.
В нашем исследовании для изучения иммунопатогенеза серонегативной миастении была исследована сыворотка крови 11 серонегативных больных на наличие АТ к MuSK. При этом для определения специфичности АТ к MuSK в качестве маркера серонегативной миастении, изучение концентрации АТ к МuSK прошли 13 серопозитивных больных миастенией, титр АТ к АХР у которых был наиболее высоким в серопозитивной группе (средние значения 20,44 ± 3,87 Нмоль /л, при норме не более 0,5 Нмоль/л).
Проведенные нами исследования показали значительное повышение титра АТ к MuSK у 45,5% больных с серонегативной миастенией (средние значения составили 1,1±0,5 Нмоль/л, при норме менее 0,05 Нмоль/л, максимальные значения 1,36 Нмоль/л, минимальные 0,13 Нмоль/л). При этом серопозитивные больные были MuSK-негативными во всех случаях.
Таким образом, мы получили собственные данные, что АТ к MuSK являются достаточно специфичным маркером серонегативной миастении. Вместе с тем, выявление этих антител не у всех серонегативных больных позволяло предполагать о неоднородности патогенеза этой формы заболевания. Клинические отличия MuSK-позитивных и MuSK-негативных групп СН миастении проявлялись в чувствительности к антихолинэстеразным препаратам. Так у серонегативных больных с отсутствием АТ к MuSK отмечалась слабая или частичная обратимость нарушений нервно-мышечной передачи на прозерин. При MuSK-позитивной серонегативной миастении клиническая и электрофизиологическая реакции на прозерин были негативными, наиболее часто обнаруживались повторные М-ответы, рис.6.
Рис. 6(А)
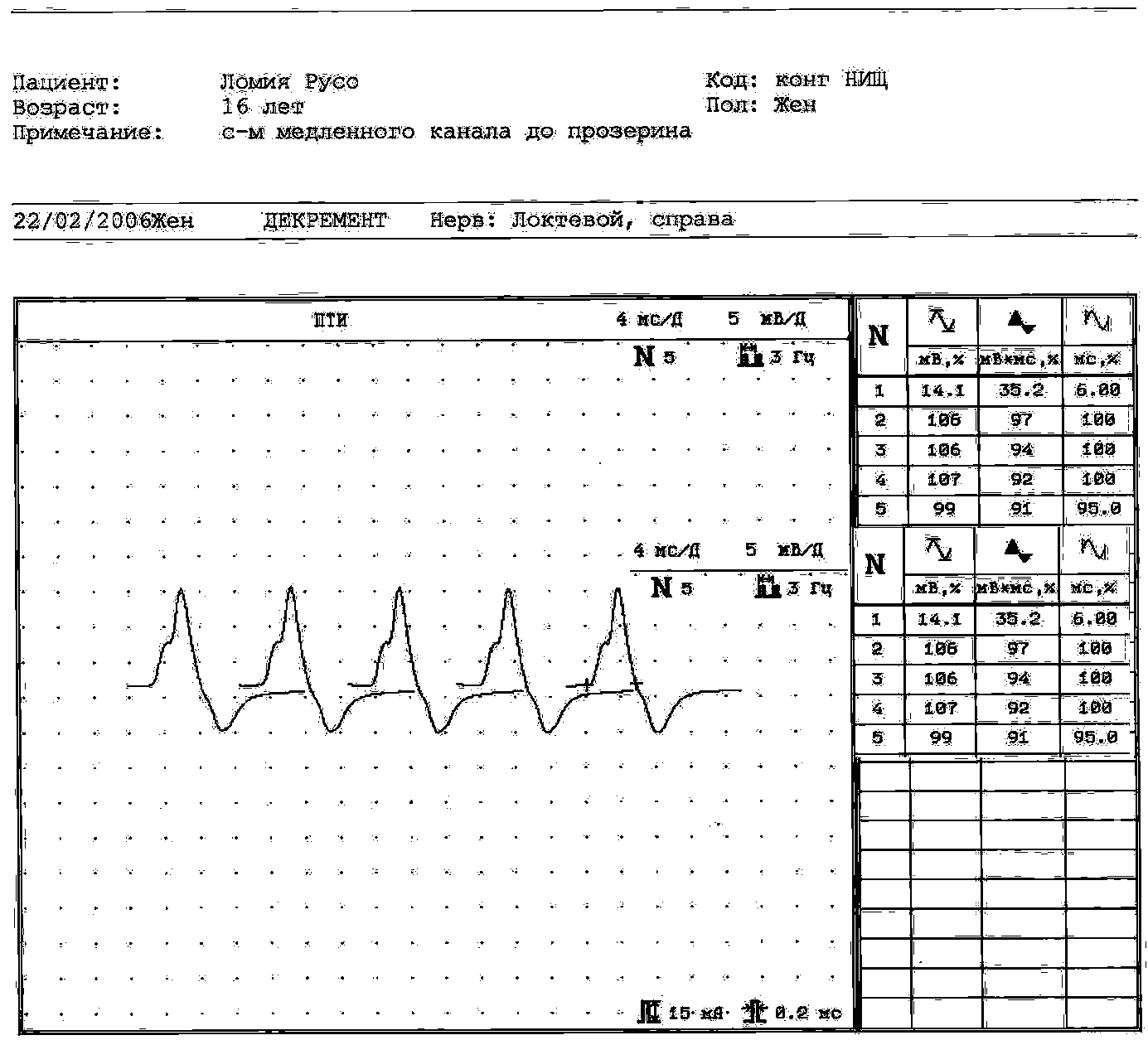
Рис.6(Б)
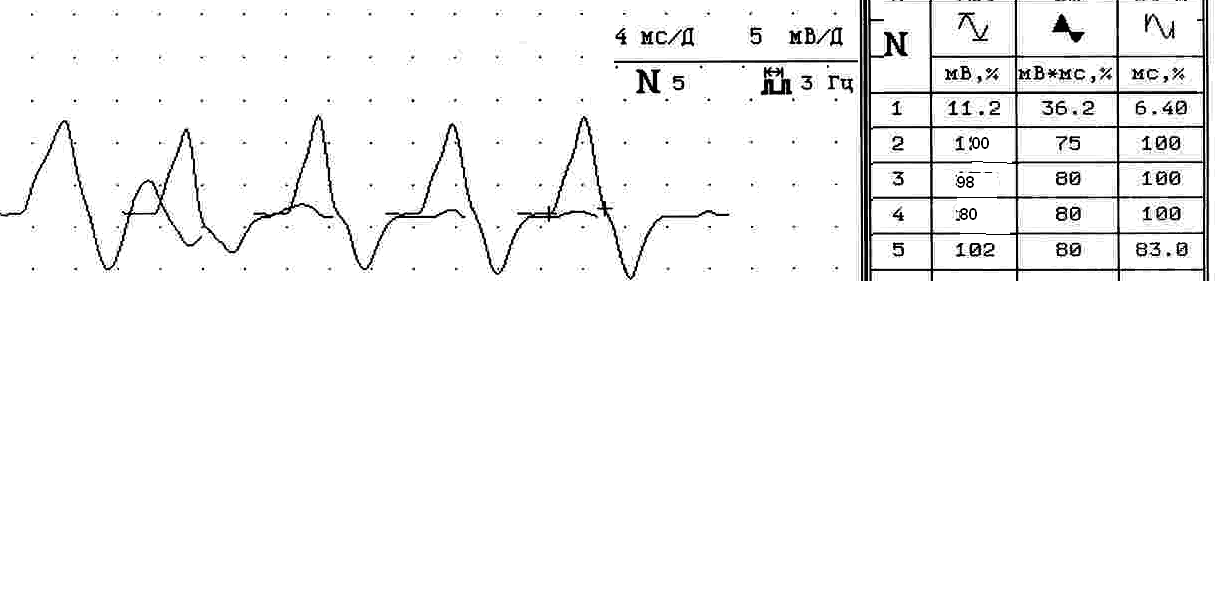
Рис 6. Отсутствие (А) и появление на фоне прозерина (Б) повторного компонента в мышце, отводящей мизинец кисти у больной Л., 18 лет серонегативная MuSK-позитивная миастения. Титр АТ к MuSK 1.27 Нмоль/л при норме менее 0.005 Нмоль/л.
6(А). При стимуляции частотой 3 имп/с серией из 5 стимулов в фоновом исследовании амплитуда М-ответа составляет 14,1 мв, площадь 35,2 мвмс, длительность 6,0 мс.
6(Б). Та же мышца через 40 минут после введения прозерина: появляется дополнительный компонент М-ответа, декремент которого –71%. Амплитуда основного компонента М-ответа на фоне прозерина уменьшается до 11,2 мв, площадь увеличивается до 36,2 мвмс, а длительность увеличивается до 6,4 мс. В мышце отмечалась Н-холинергическая реакция на фоне прозерина (фасцикуляции), что отразилось на нестабильности М-ответов в серии.
Основные модели нарушений нервно-мышечной передачи при трех типах патологии синапса у больных миастенией, выявляемые при ритмической стимуляции частотой 3 имп/c и в период постактивационного облегчения (ПАО), представлены на рисунке 7.
I тип патологии синапса (больная К., 22 года)
Фон ПАО

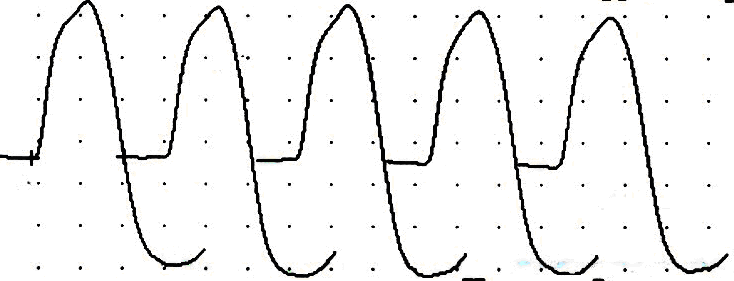
Амплитуда 7,4 мВ, декремент -40% Амплитуда 7,5 мВ, декремент -5%