
Тезисы лекций и стендовых докладов
| Вид материала | Тезисы |
- Тезисы докладов, 4952.24kb.
- Тезисы докладов, 3726.96kb.
- «Актуальные вопросы кардиологии», 58.59kb.
- Тезисы докладов, 1225.64kb.
- Правила оформления тезисов докладов Тезисы докладов предоставляются в электронном виде, 22.59kb.
- «Симпозиум по ядерной химии высоких энергий», 1692.86kb.
- Требования к тезисам докладов, 16.83kb.
- Тезисы докладов научно-практической, 6653.64kb.
- Правила оформления материалов объем тезисов не должен превышать 1 страницы. Текстовый, 24.85kb.
- Образец оформления стендовых докладов, 4.6kb.
ПОЛЯРНЫЙ ЭФФЕКТ В РЕАКЦИЯХ ПОЛИМЕРИЗАЦИИ И СОПОЛИМЕРИЗАЦИИ МОНОМЕРОВ
Денисова Т.Г.
Институт проблем химической физики РАН,
Черноголовка, Россия, e-mail: denisova@icp.ac.ru
В рамках модели реакции радикального присоединения как суперпозиции трех потенциальных кривых, описывающих потенциальную энергию валентного колебания связи как функцию амплитуды ее колебания, проанализированы экспериментальные данные (константы скорости) радикальной полимеризации и сополимеризации полярных мономеров. Такой подход позволяет по экспериментальным данным вычислить энергию активации термонейтрального аналога рассматриваемой реакции и разделить вклад энтальпии реакции, триплетного отталкивания и полярного взаимодействия в энергию активации присоединения мономера к растущему макрорадикалу.
Ниже приводится пример такой оценки для реакций полимеризации ряда полярных мономеров. В качестве реакции сравнения взята реакция продолжения цепи при полимеризации этилена. Вклад энтальпии в энергию активации вычислялся как разность:
DEH = Е – Е0. Вклад взаимодействия полярных групп вычислялся как разность между энергиями термонейтральных реакций присоединения полярного мономера и этилена: DEm = E0 (CH2=CHX) – E0(CH2=CH2).
| Полимер | –DH | E | E0 | DEH | DEm |
| | кДж/моль | ||||
| CH2=CH2 | 93.5 | 37.4 | 119.2 | –81.8 | 0.0 |
| CH2=CHOAc | 114.9 | 30.4 | 118.4 | –88.0 | –0.8 |
| CH2=CHCO2Me | 96.3 | 31.0 | 114.5 | –83.5 | –4.7 |
| CH2=CMeCO2Me | 98.5 | 33.0 | 108.1 | –75.1 | –11.0 |
| CH2=CHCN | 119.7 | 20.2 | 111.0 | –90.8 | –8.2 |
| CH2=CHCl | 120.7 | 24.0 | 116.0 | –92.0 | –3.2 |
Как видно из приведенных данных, полярный эффект снижает энергию активации и в ряде случаев весьма значительно (на 8 ¸ 11 кДж/моль). После энтальпии реакции это самый существенный фактор, влияющий на энергию активации индивидуальной реакции.
КОМБИНАТОРНАЯ СИММЕТРИЯ ТОПОЛОГИЧЕСКИХ СВЯЗЕЙ И ЭФФЕКТ СЛИЯНИЯ ЗВЕНЬЕВ И МОД ПРИ ВЯЗКОУПРУГОЙ РЕЛАКСАЦИИ РАЗВЕТВЛЕННЫХ ОЛИГОМЕРОВ
Дубовицкий В.А., Иржак В.И.
ИПХФ РАН, г. Черноголовка, Россия, dubv@icp.ac.ru
Рассматривается линейная модель релаксации, в которой макромолекулы олигомеров представляются как система легких упруго связанных частиц, движущихся в вязкой среде. Нередко в структуре графа топологических связей разветвленных макромолекул присутствует симметрия относительно перестановок. Например, если переставить и перенумеровать от "корня" две ветви правильной звездообразной макромолекулы, то вид уравнений движения сохранится. Такая же ситуация имеет место для сверхразветвленной макромолекулы с бинарным графом связей: можно переставлять ветви, исходящие из общего узла и при этом уравнения движения останутся неизменны.
Наличие единой перестановочной симметрии в топологических связях, модулях упругости и коэффициентах трения и начальных данных приводит к тому, что группы узлов и связей "сливаются" и движутся как единое целое. Это вытекает из следующего: если коэффициенты трения и упругой связи, начальные условия звеньев инвариантны относительно некой перестановки - симметрии, то и компоненты решения уравнений релаксационного движения инвариантны относительно этой перестановки. Из этого простого предложения вытекает, что звенья попадающие в непересекающиеся циклические классы, порожденные перестановкой-симметрией совпадают в ходе релаксационного движения.
В итоге движение исходной макромолекулы точно описывается сокращенной агрегированной системой уравнений движения, которая получается суммированием параметров трения, связи слившихся звеньев. Поскольку число релаксационных мод в разложении целевой функции-модуля (например, напряжения) связано с размерностью системы уравнений движения, из-за эффекта слияния косвенно сокращается и число ненулевых компонент экспоненциального разложения, т.е. релаксационный спектр. В докладе показано, что следствием эффекта слияния является эквивалентность релаксации свернутых звезд и гиперразветвленных молекул релаксации неоднородных линейных цепей, размер которых не превосходит удвоенной длины ветвей и числа поколений, причем явно указаны параметры этих цепей.
Работа выполнена при поддержке РФФИ, проекты 07-01-00588, 07-03-00104.
КРИТИЧЕСКАЯ КОНВЕРСИЯ ПРИ ЦИКЛОТРИМЕРИЗАЦИИ.
Иржак Т.Ф., Иржак В.И.
Институт проблем химической физики РАН
Черноголовка, Россия, irzhak@icp.ac.ru.
Реакция циклотримеризации, которая характерна для изоцианатов, производных циановой кислоты и алкинов, с кинетической точки зрения представляет собой особый случай поликонденсационных процессов, ибо рост полимерной цепи осуществляется за счет взаимодействия не двух, как обычно, а трех функциональных групп. Вне зависимости от химических особенностей механизма реакции ее можно представить как тримолекулярную в некотором фиктивном времени.
Предложена общая схема циклотримеризации моно- и бифункциональных олигомеров без учета эффекта замещения, но с учетом разной реакционной способности функциональных групп с допущением аддитивности свободной энергии активации, т.е. в предположении, что константу скорости реакции А+В+С kabc можно выразить как kabc=kakbkc. Приведены результаты расчетов для зависимости величины критической конверсии aС от состава реакционной смеси и относительной реакционной способности функциональных групп. Обнаружен экзотический характер зависимости, а именно, уменьшение величины критической конверсии от концентрации обрывателя цепи развития сетки, монофункционального агента, в случае его низкой реакционной способности, обусловленный тем, что сетка формируется, главным образом, за счет бифункционального агента, и aС падает из-за снижения его доли.
Учет эффекта замещения ведет к существенному усложнению схемы реакции вследствие появления дополнительной константы скорости k3, характеризующей реакционную способность второй функциональной группы В®C, если первая прореагировала. В связи с этим приходится различать не четыре типа реакций, как выше, а десять.
Приведены результаты расчетов, показывающих зависимость величины критической конверсии aС от состава реакционной смеси и величины эффекта замещения, т.е. относительной реакционной способности функциональных групп С. Показано, что положительный эффект замещения при относительно невысоком содержании монофункционального компонента понижает величину критической конверсии, тогда как отрицательный ведет к некоторому повышению aС.
ПОЛЯРНЫЕ ЭФФЕКТЫ В РЕАКЦИЯХ ПРИСОЕДИНЕНИЯ ФЕНОКСИЛЬНЫХ И НИТРОКСИЛЬНЫХ РАДИКАЛОВ К ДВОЙНЫМ СВЯЗЯМ ОЛИГОМЕРНЫХ ЭФИРОВ (МЕТ)АКРИЛОВОЙ КИСЛОТЫ
Капранов В.А., Сирик А.В., Плисс Е.М.
Ярославский государственный университет им. П.Г. Демидова,
г. Ярославль, Россия, pliss@uniyar.ac.ru
Изучена кинетика присоединения 2,4,6-три(трет.бутил)феноксиль-ного (PhO.) и 4,4-диметоксидифенилнитроксильного ([>NO.) радикалов к p-связям олигоэфиров (мет)акриловой кислоты и полиолов (метод ЭПР). Расчет констант скорости по экспериментальным данным проводили по оригинальной оптимизационной программе. Расчет энтальпии присоединения радикалов осуществлялся квантовохимически (методом АМ1).
Изменение концентрации PhO· { Ar, ([PhO·]=10-3 - 10-4 моль/л, [>C=C<]=0.5-4,5 моль/л)} хорошо описывается зависимостью kPhO·t=ln([PhO·]0/[PhO·])/ Кинетика расходования >NO×) в принятых экспериментальных условиях {(Ar, [>NO·]=10-3-10-4 моль/л, [
 ]=0.5-4,5 моль/л} также описывается уравнением: ln([>NO·]0/[>NO·])=k NO· t.
]=0.5-4,5 моль/л} также описывается уравнением: ln([>NO·]0/[>NO·])=k NO· t.Полученные данные (Таблица) свидетельствуют, что по мере увеличения числа (мет)акрилатных групп в олигомере значения kPhO· и kNO· (в расчете на одну p-связь)существенно падают по сравнению с моноэфирами (бутилакрилат и метилметакрилат).
| 105kPhO· и108 (kNO·, л/( моль.с), 353К | |||||||
| Олиго-мер | БА (1) | НГДА (2) | ТПТА (3) | ПЭТА (4) | ММА (1) | ТПТМ (3) | ПЭТМ (4) |
| PhO. | 5,.1 | 4.8 | 0,7 | 0,6 | 1,1 | 0,4 | 0,3 |
| >NO· | | | | | 2,2× | 0,3 | 0,2 |
В скобках – число (мет)акрилатных групп в олигоэфире
Данная феноменология обусловлена эффектом мультидипольного взаимодействия (Е.Т. Денисов), обнаруженным ранее для присоединения RO2×, ROOH и O2 к p-связям данных олигоэфиров (Е.Т. Денисов, Е.М. Плисс с сотр.). Существенная разница в значениях kPhO· и kNO· (три порядка) обусловлена различиями в термохимии присоединения PhO· и >NO..
ВНУТРИМОЛЕКУЛЯРНАЯ ПОДВИЖНОСТЬ ЦЕПЕЙ
ПОЛИ(ФТОР)АЛКИЛМЕТАКРИЛАТОВ
Ладинин М.Е., Ильин А.А., Соловьев М.Е.
Ярославский государственный технический университет,
г. Ярославль, ladininme@ystu.ru
В последнее время установлена чувствительность кинетики полимеризации высших алкил(мет)акрилатов к введению малых добавок (1-5%) других алкил(мет)акрилатов [Королев Г.В., Ильин А.А., Могилевич М.М., Евплонова Е.С., Грачев В.П. // Высокомолек. соед.- А.- 2003. Т. 45. № 6. С. 892.] с той же реакционноспособностью. Так как мономер-добавка расходуется в процессе полимеризации как и основной мономер, встраиваясь в растущую полимерную цепь, то эти кинетические эффекты, вероятно, должны приводить к структурным изменениям синтезируемого полимера и в конечном счете сказываться на его свойствах.
Цель данной работы – моделирование внутримолекулярной подвижности полимерных цепей полидодецилметакрилата (ПДДМА), получаемых в отсутствии и присутствии 4% мол. бутилметакрилата (БМА), а также полимерных цепей полидодекафторгептилметакрилата (ПДФГМ), получаемых в отсутствии и присутствии 4% мол. тетрафторпропилметакрилата (ТФПМ).
Объектами для компьютерного моделирования служили 50-звенные фрагменты полимерных цепей ПДДМА (ПДФГМ) и ПДДМА, (ПДФГМ), у которых 17 и 34 звеньями являются звенья БМА (ТФПМ). Для моделирования внутримолекулярной подвижности фрагментов полимерных цепей использовался метод молекулярной динамики – проводилось численное интегрирование уравнений движения Ньютона с использованием алгоритма Беемана [Хеерман Д.В. Методы компьютерного эксперимента в теоретической физике. – М: Наука, 1990. – 176 с.].
Статистический анализ динамического поведения фрагментов полимерных цепей ПДДМА (ПДФГМ) и ПДДМА-БМА (ПДФГМ-ТФПМ) в течение 1000 пс после достижения «тепловой релаксации» при заданных температурах (в диапазоне 50 – 300 К) показал, что замена двух ДДМА (или ДФГМ) звеньев в 50-звенных полимерных фрагментах ПДДМА (ПДФГМ) на два звена БМА (ТФПМ) приводит к значительному увеличению флуктуации средних расстояний между конечными атомами углерода основных полимерных цепей и косинусов двухгранных углов в местах локализации инородных звеньев.
МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ КИНЕТИКИ РАДИКАЛЬНО-ЦЕПНОЙ ПОЛИМЕРИЗАЦИИ С УЧЕТОМ АССОЦИАЦИИ РЕАГЕНТОВ
Смирнов Л.П., Карнаух Г.Е., Кулагина Т.П.
Институт проблем химической физики РАН,
Черноголовка, Россия, tan@icp.ac.ru
Олигомеры, а также мономеры, образуют ассоциаты (домены, кластеры и т.д.), причем реакционная способность последних, как правило, заметно отличается от таковой для исходных реагентов [С.М.Межиковский, А.Э.Аринштейн, Р.Я.Дебердеев. Олигомерное состояние вещества М.; Наука. 2005]. Между тем, надмолекулярная структура реагентов обычно не принимается во внимание при расчете кинетических параметров по экспериментальным данным о радикально-цепной полимеризации, и, таким образом, расчетные константы скорости являются по существу эффективными характеристиками, зависящими как от истинной константы скорости полимеризации, так и от константы равновесия процесса ассоциации. Это обстоятельство не дает возможность выявить детальный механизм и определить истинные кинетические характеристики радикально-цепной полимеризации в жидкой фазе. Это, в свою очередь, в какой-то степени препятствует развитию теоретических основ управления кинетикой этих реакций за счет внешних физических воздействий. Так, в частности, ни одна из теоретических моделей, описывающих влияние звука на скорость химической реакции, не учитывает изменение надмолекулярной структуры реагентов под воздействием звука [М.А.Маргулис. Звукохимические реакции и сонолюминесценция. М: Химия. 1986].
В работе предложена новая математическая модель кинетики радикально-цепной полимеризации, включающая, наряду со стадиями инициирования (с образованием радикальной пары и выхода радикалов из клетки), роста и обрыва кинетической цепи, стадий образования и распада ассоциатов реагентов. Анализ системы кинетических уравнений проводился численным методом. В приближении квазистационарности концентрации радикалов в аналитическом виде получены соотношения, связывающие скорость полимеризации с константой равновесия ассоциации реагентов. Исследовано влияние напряжений, создаваемых акустическим воздействием, на скорости стадий распада ассоциатов, образования радикальной пары и рекомбинацию радикалов. Показано, что скорость процесса полимеризации определяется в основном величиной амплитуды акустических колебаний.
Синтез олигомеров.
Новые функциональные олигомеры. Получение и свойства
МЕХАНИЗМ ОКИСЛЕНИЯ ОЛИГОМЕРОВ ДЕЦЕНА-1
Базанов Т.А., Петров Л.В., Психа Б.Л., Харитонов В.В.
Институт проблем химической физики РАН,
г. Черноголовка, Россия. E-mail: psi@icp.ac.ru
На основе разработанного в ИПХФ РАН метода катионной олигомеризации олефинов создана технология и построен завод по производству синтетических олигодеценовых масел. С целью повышения окислительной стабильности производимых масел ведется работа по исследованию процесса окисления основных продуктов олигомеризации децена-1. В настоящей работе проведено кинетическое исследование механизма окисления фракции ПАО-2, более чем на 90% состоящей из димеров децена-1. При 120, 130 и 140°С изучены кинетические закономерности автоокисления и инициированного окисления исходного и гидрированного образцов ПАО-2, идентифицированы ключевые реакции в механизме процесса и определены численные значения кинетических параметров. Установлено, что причиной высокой окисляемости исходного олигомера, кроме наличия в образце двойных связей и гидропероксидов, являются большие значения скорости самопроизвольного зарождения радикалов и константы скорости распада гидропероксидов на радикалы, что связано с наличием в исследуемом образце различных примесей, в том числе каталитического характера.
Показано, что в результате гидрирования исходного ПАО-2 происходит положительное изменение всех параметров окисления, что и приводит к увеличению окислительной стабильности олигомера.
Проведено сравнительное исследование механизма окисления образца ПАО-2, полученного по измененной технологии. Показано, что от технологии производства зависят как качественные, так и количественные показатели окислительной стабильности получаемого олигомера. Внесены необходимые дополнения в механизм окисления нового образца и определены значения соответствующих параметров.
С целью повышения окислительной стабильности исходных, негидрированных продуктов олигомеризации проведено исследование процесса окисления образца ПАО-2 в присутствии антиоксидантов класса пространственно-затрудненных фенолов и ароматических аминов. Показано, что кинетические закономерности ингибированного окисления ПАО-2 соответствуют классическим представлениям теории жидкофазного окисления, и рассмотренные ингибиторы могут быть использованы для защиты от окисления первичных продуктов олигомеризации в период их временного хранения.
ОЛИГОДИЕНЫ С КОНЦЕВЫМИ ФУНКЦИОНАЛЬНЫМИ ГРУППАМИ. СТРОЕНИЕ И СВОЙСТВА.
Баранцова А.В., Грищенко В.К., Бусько Н.А., Кочетова Я.В.
Институт химии высокомолекулярных соединений НАН Украины
Киев, Украина, E-mail: oligomer8@bigmir.net
Изучены закономерности синтеза и свойства олигодиенов с концевыми функциональными группами: гидразидными, ацилгидразонными, уретанизоцианатными, оксадиазолинил-карбаматизоцианатными.
Показано влияние образования водородных связей между концевыми функциональными олигодиенов группами на их реологические характеристики, а также на физико-механические свойства полученных на их основе полимерных материалов.
При сравнении строения и реологических характеристик функциональных олигодиенов установлено: чем выше уровень водородных связей между полярными группами, тем выше их вязкость во всем интервале температур. При практически одинаковых строении олигомерной цепи и молекулярной массе, олигодиены с различными функциональными группами значительно отличаются по вязкости, которая зависит от числа функциональной групп и силы межмолекулярного взаимодействия одной группы.
Аналогичные влияния происходят в полимерах на основе олигомеров с концевыми функциональными группами. В результате ИК-спектроскопического исследования установлено, что количество водородносвязанных групп в свежеприготовленных полимерах отличается. Симбатно с количеством водородносвязанных групп в растворе полимеров происходят ассоциативные процессы, что выражается в росте вязкости раствора полимера в неполярных растворителях. После удаления растворителя линейные полимеры не растворимы в обычно применяемых растворителях, в том числе и в тех, в которых получены, происходит физическое гелеобразование (полная потеря текучести).
Методом МУРР показано, что водородные связи являются основной движущей силой ассоциации, следовательно, и сегрегации блоков различной природы сегментированных полимеров.
Олигомеры металлокомплексов
тетра-(2,3-тиофено)порфиразина
Браханова О.О., Корженевский А.Б., Койфман О.И.
Ивановский государственный химико-технологический университет,
Институт химии растворов РАН, Иваново, Россия, kobuko@isuct.ru
Методом темплатной полициклотетрамеризации 2,3-дициантиофена и 2,3,4,5-тетрациантиофена в присутствии солей соответствующих металлов и каталитического количества ацетата аммония синтезированы олигомерные металлокомплексы тетра(2,3-тиофено)порфиразина – тиаизолога фталоцианина.
Структура олигомеров определяется соотношением в реакционной смеси ди– и тетрафункционального мономеров.
П
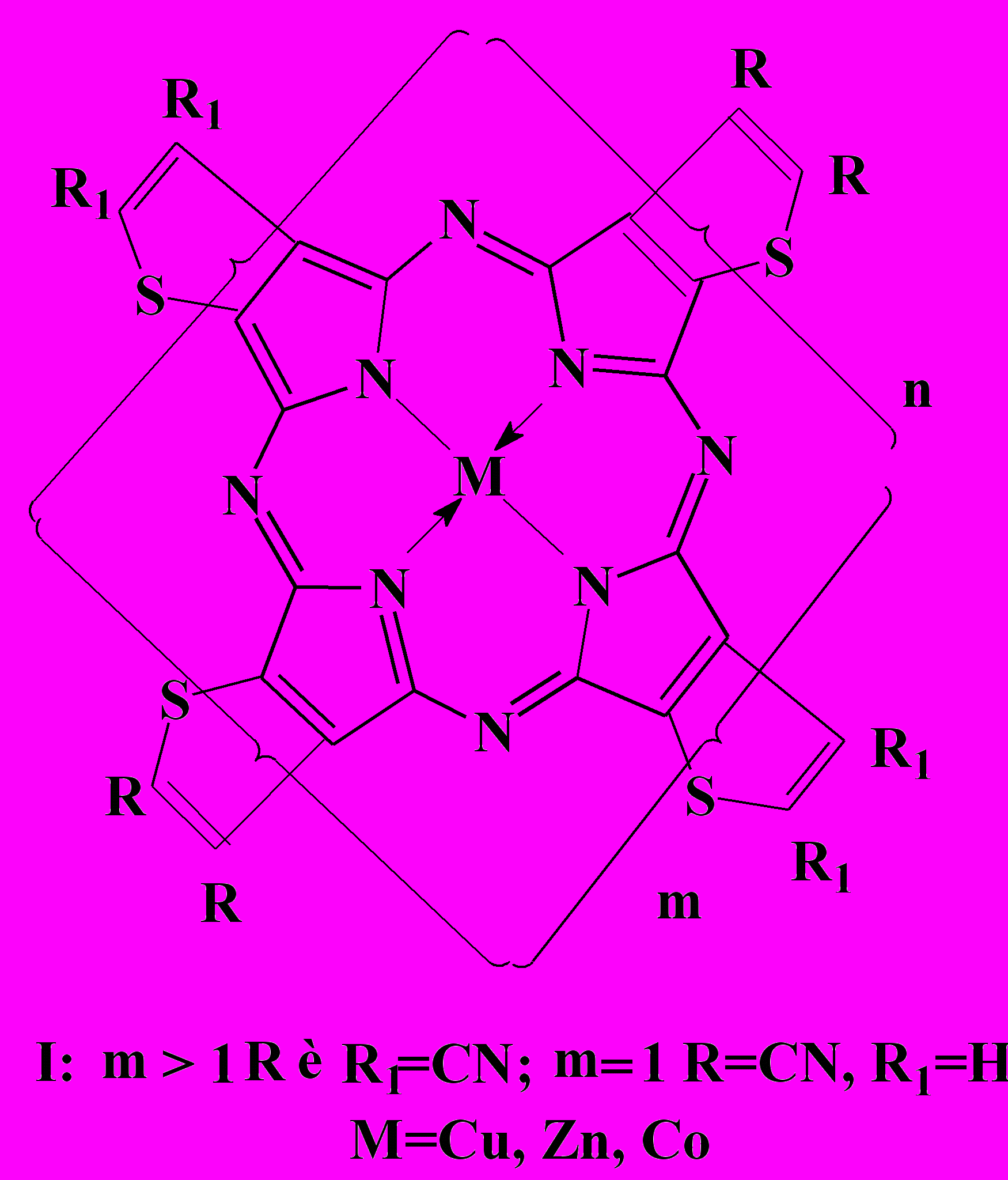 олученные олигомерные металлокомплексы имеют ряд уникальных особенностей:
олученные олигомерные металлокомплексы имеют ряд уникальных особенностей:- жесткая плоскостная структура тетразапорфинового остова элементарного звена;
- возможность введения в состав олигомера различных металов;
- наличие единой π-электронной система всей макромолекулы;
- наличие в составе макролиганда азатиакраун-полостей и подандов нескольких типов, обладающих различной координационной активностью;
- возможность химической модификации олигомера взаимодействием по атомам серы;
Идентификация структуры олигомеров по электронным спектрам поглощения, данным элементного анализа и содержанию концевых групп показала, что полученные соединения представляют собой олигомеры со степенью полимеризации не более 6. Предельные размеры молекул, очевидно, обусловлены термодинамическими причинами и, в частности, потерей ароматичности за счет сильного искажения плоскостной структуры молекулы по мере увеличения ее размеров.
Перспективы использования в качестве катализаторов, комплекситов, сенсоров, полупроводников, материалов для нелинейной оптики и основы супрамолекулярных рецепторов делают полученные олигомеры интересными не только в научном плане.
СИНТЕЗ И СВОЙСТВА РАЗВЕТВЛЕННЫХ АЛКИДУРЕТАНОВЫХ БЛОКСОПОЛИМЕРОВ
Бубнова А.С.
Институт химии высокомолекулярных соединений НАН Украины, Киев, Украина, E-mail: asbubnova@ukr.net
Синтезированы разветвленные блоксополимеры на основе алкидных смол и функционализированных изоцианатсодержащих олигомеров различной химической природы, полученных с использованием алифатических, ароматических изоцианатов и растительных масел, соотношение компонентов было различным. Синтез осуществлялся в 2 стадии: первая стадия – приготовление функционализированных изоцианатсодержащих олигомеров, вторая – синтез алкидуретановых блоксополимеров путем химической модификации наполненных и ненаполненных алкидных смол изоцианатными олигомерами.
Изучение кинетических закономерностей процесса образования блоксополимеров показало, что реакционная способность функционализированных изоцианатных олигомеров по отношению к алкидным смолам изменяется в ряду: ароматический триизоцианат>триизоцианат на основе растительного масла>алифатический триизоцианат, что позволяет выбрать оптимальный состав разветвленных блоксополимеров для синтеза материалов, обладающих высокими защитными и декоративными свойствами.
Установлено, что для обеспечения высоких технологических характеристик композиционных полимерных материалов на основе изоцианатных олигомеров и алкидных смол, реакции образования разветвленного блоксополимера необходимо обрывать после 50-60% превращения изоцианатных групп модификатора. Для чего вводились соединения, имеющие активную аминную или гидроксильную группу, взаимодействующую с изоцианатом. Катализаторы окислительной полимеризации (сиккативы) являются в то же время и катализаторами реакции уретанообразования при модификации алкидных смол.
Разветвленные алкидуретановые блоксполимеры, были охарактеризованы методами ИК-спектроскопии, эбулиоскопии и гель-проникающей хроматографии. Увеличение молекулярной массы в 3 раза подтверждает образование разветвленной структуры модифицированного продукта в отличие от исходного алкида.
СИНТЕЗ ОЛИГОМЕРОВ С КОНЦЕВЫМИ ФУНКЦИОНАЛЬНЫМИ ГРУППАМИ МЕТОДОМ ФОТОИНИЦИИРОВАННОЙ РАДИКАЛЬНОЙ ПОЛИМЕРИЗАЦИИ
Бусько Н.А., Грищенко В.К., Баранцова А.В., Фальченко З.В.
Институт химии высокомолекулярных соединений НАН Украины,
г. Киев, Украина, e-mail: oligomer@bigmir.net
Исследованы закономерности синтеза мономерных азоинициаторов радикальной полимеризации с концевыми ацилгидразонными группами, которые хорошо растворимы в углеводородных растворителях. Устиновлено, что константы скорости распада азоинициаторов увеличиваются с увеличением полярности растворителей, что связано с наличием у них функциональных групп, способных к сольватации растворителем. Изучена кинетика фотодиссоциации азоинициаторов и показано, что наиболее эффективным из них является 2,2'-азо-бис-изобутирогидразон циклогексанона, который имеет наиболее высокий молярный коэффициент экстинкции, константу распада и хорошо растворим в углеводородных растворителях.
Синтезированные азоацилгидразоны могут быть использованы не только как термоинициаторы, но и как фотоинициаторы для синтеза олигомеров, что дает им определенные преимущества по сравнению с использованием их только в качестве термоинициаторов. В случае фотоинициированной полимеризации оказывается возможным поддерживать необходимую температуру, которой определяется соотношение kp/ko1/2 и, независимо, изменением мощности излучения (мощность УФ-лампы, расстояния, наличие фильтров) изменять скорость инициирования.
На основе диеновых и винильных мономеров с применением синтезированных нами азоинициаторов получены в мягких условиях (температура 293-298 К) в растворе и в массе при фотоинициируемой полимеризации, и более жестких условиях (температура 368-370 К и высокое давление) при термоинициируемой полимеризации, реакционноспособные олигомеры.
СИНТЕЗ ОЛИГОМЕРОВ НА ОСНОВЕ СТИРОЛА С ИСПОЛЬЗОВАНИЕМ МЕТАЛЛОСОДЕРЖАЩИХ КОМПЛЕКСОВ КАК РЕГУЛЯТОРОВ РОСТА ЦЕПИ
Валетова Н.Б., Ильичев И.С., Гришин Д.Ф.+
Научно-исследовательский институт химии Нижегородского государственного университета им. Н.И.Лобачевского
Н.Новгород, Россия, е-mail: grishin@ichem.unn.ru
В плане разработки новых инициирующих систем для синтеза олигомеров нами изучена сополимеризация стирола со стиролхромтрикарбонилом в присутствии никелевой каталитической системы NiBr2(PPh3)2/Zn/Ph-Br в среде ацетонитрила и пиридина.
Попытка получить стильбенхромтрикарбонил в результате реакции сочетания между бромбензолом и стиролхромтрикарбонилом при 650С в присутствии NiBr2(PPh3)2/Zn/Ph-Br не привела к желаемому результату [Ilitchev I.S., Tikhonova Y.V., Grishin D.F. Abstracts of “Modern trends in organoelement and polymer chemistry”, Moscow, Russia. 2004. P. 146], хотя известно, что сочетание бромбензола со стиролом в присутствии исследуемого никелевого катализатора приводит к стильбену [Lebedev S.A., Lopatina V.S., Petrov E.S., Beletskaya I.P. // J. Organometal. Chem. 1988. V. 44. № 3. P. 253-259]. При добавлении стиролхромтрикарбонила к бромбензолу и стиролу (мольное соотношение 1:1:2) в присутствии никелевой каталитической системы NiBr2(PPh3)2/Zn/Ph-Br (мольное соотношение 0,05:3:1) в среде ацетонитрила и пиридина обнаружено, что в качестве основного продукта был получен сополимер стирола со стиролхромтрикарбонилом с выходом 44%. Продукт кросс-сочетания - стильбенхромтрикарбонил выделен не был. Установлено, что с увеличением концентрации хромсодержащей добавки с 20 до 50 мол. % при полимеризации стирола в присутствии NiBr2(PPh3)2/Zn/Ph-Br наблюдается равномерное снижение среднечисленной молекулярной массы полимера с 5500 до 3000 при проведении полимеризации в течение 4,5 ч при 650С. Таким образом, каталитическая система на основе бромбензола и дибромида бис(трифенилфосфин)никеля в присутствии цинковой пыли в ацетонитриле и пиридине способна эффективно инициировать синтез металлосодержащих олигомеров стирола и стиролхромтрикарбонила.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (грант № 05-03-32688).
Соотношение реакций поликонденсации и дегалогенирования при синтезе Халькогенсодержащих олигомеров
Вшивцев В.Ю., Леванова Е.П., Грабельных В.А., Елаев А.В.
Сухомазова Э.Н., Руссавская Н.В., Корчевин Н.А.
Иркутский институт химии им. А.Е. Фаворского СО РАН,
Иркутск, Россия. E-mail: venk@irioch.irk.ru
Халькогенсодержащие олигомеры обычно получают поликонденсацией полиэлектрофилов с халькогенид-анионами Yn2- (Y=S, Se, Te; n=1-4). Для вицинальных диэлектрофилов, например, для 1,2-дигалогенэтанов, реакция с K2Sn протекает по схеме поликонденсации, а с селенидами и теллуридами наблюдается дегалогенирование с выделением этилена и халькогенов в свободном состоянии [Н.В. Руссавская, В.А. Грабельных, Е.П. Леванова, Э.Н. Сухомазова, Л.В. Клыба, Е.Р. Жанчипова, А.И. Албанов, А.А. Татаринова, А.В. Елаев, Э.Н. Дерягина, Н.А. Корчевин, Б.А. Трофимов. ЖОрХ. 2006. Т. 42. Вып. 5. С. 672-678].
Нами исследован широкий ряд 1,2-диэлектрофилов (I) (1,2-дигалогенэтаны, эпихлоргидрин, 2,3-дибромпропанол-1, 1,3-дихлорпропанол-2, 2,2’-дихлордиэтиловый эфир, этилен-хлоргидрин и др.) в реакциях с элементными халькогенами в основно-восстановительных системах, которые активируют халькогены с образованием полихалькогенид-анионов Yn2-.
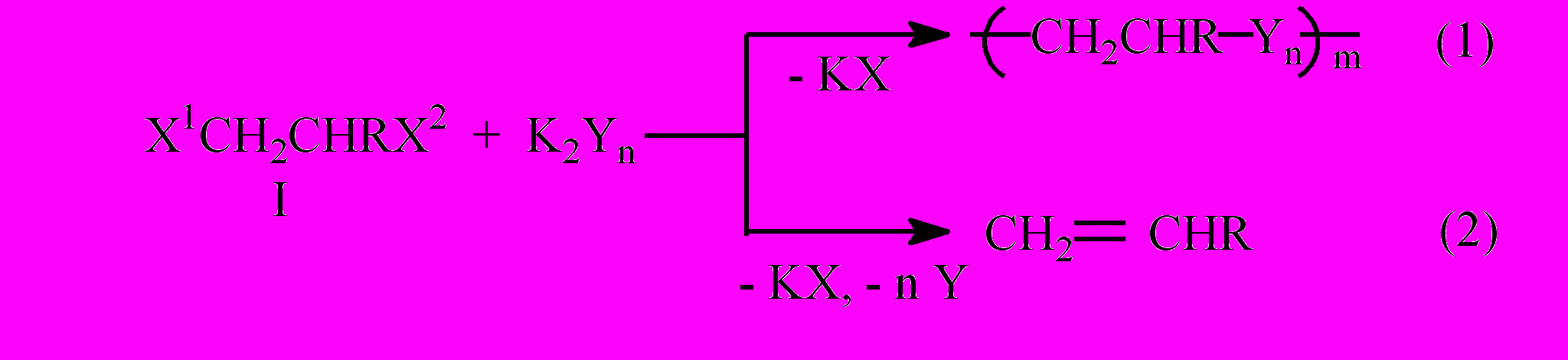
Выявлена роль характера элемента Y, заместителя X2 , типа основно-восстановительной системы, условий реакции на соотношение процессов (1) и (2).
Халькогенсодержащие олигомеры на основе хлорекса
Грабельных В.А., Вшивцев В.Ю., Елаев А.В., Леванова Е.П.,
Сухомазова Э.Н., Руссавская Н.В., Корчевин Н.А.
Иркутский институт химии им. А.Е. Фаворского СО РАН,
Иркутск, Россия. E-mail: venk@irioch.irk.ru
Олигомеры, содержащие серу, селен или теллур, обладают важными электрофизическими и другими полезными свойствами [Kishore K., Ganesh K. Adv. Polym. Sci. 1995. V. 121. P. 81].
Мы исследовали возможность синтеза халькогенсодержащих олигомеров с использованием β,β’- дихлордиэтилового эфира (хлорекса), элементных халькогенов и восстановительных систем на основе гидразина.
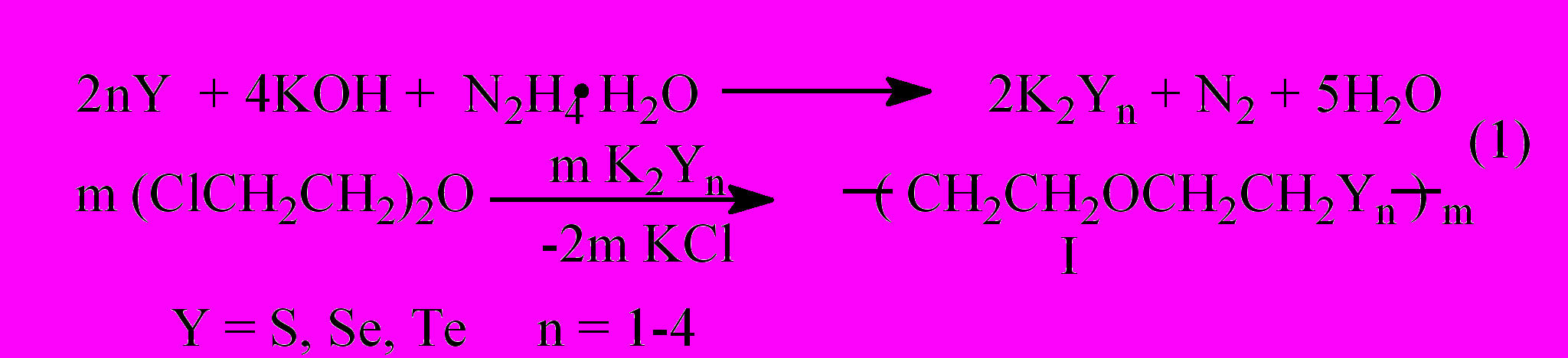
По содержанию остаточного хлора в олигомерах I оценена их молекулярная масса и величина m. При одновременном использовании двух разных халькогенов получены олигомеры II, содержащие в цепи различные халькогены.

Наряду с олигомерами в реакции (1) выделены циклические продукты III-V.

Восстановительным расщеплением олигомеров I, II и циклов IV, V получены диселенол VI, селенолотиол VII и алкильные производные VIII:
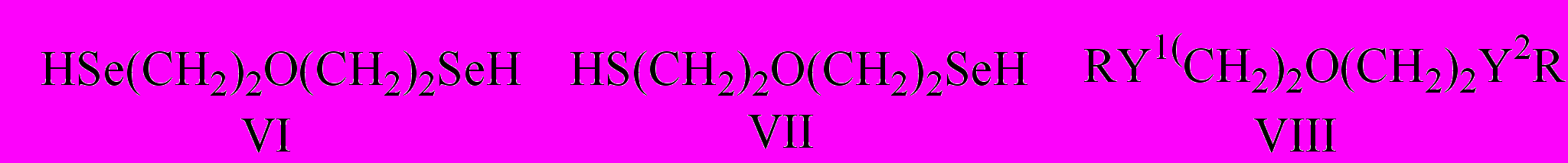
Структура синтезированых соединений III-VIII подтверждена данными ИК спектроскопии, ЯМР и масс-спектрометрии.
СИНТЕЗ ОЛИГОМЕРНЫХ БЛОКИРОВАННЫХ ИЗОЦИАНАТОВ
Джалмуханова А.С., Горбушина Г.А., Лодыгина В.П., Федотова Т.Н.,
Эстрин Я.И., Бадамшина Э.Р.
Институт проблем химической физики РАН,
г. Черноголовка, Россия, badamsh@icp.ac.ru
Известно, что изоцианаты, широко используемые в производстве различных полимеров и полимерных продуктов, в силу своей высокой реакционной способности обладают малой живучестью. Для преодоления этого недостатка, а также летучести и токсичности изоцианатов целесообразнее использовать их блокированные производные – блокированные изоцианаты и полиизоцианаты (БПИ), являющиеся продуктами частичной тримеризации диизоцианатов. Нами разработан одностадийный, технологичный способ синтеза БПИ [Патент РФ №2148061, 2000 г.], позволяющий получать как 100 %-ный преполимер, пригодный либо для формования массивных изделий, либо в качестве основы адгезивов, лаков и красок, так и готовый лак.
Задачей настоящей работы является синтез БПИ на основе диизоцианатов и блокирующих агентов различной природы и структуры с целью снижения температуры отверждения БПИ.
Синтезированы БПИ на основе толуилендиизоцианата и изофорондиизоцианата, имеющих различающиеся по реакционной способности изоцианатные группы, с использованием в качестве блокирующих агентов ε-капролактама, диэтилового эфира малоновой кислоты, диметилпиразола и 2-бутаноноксима. Установлена структура полученных БПИ методами ИК-спектроскопии и жидкостной хроматографии. Показано, что синтез БПИ на основе циклоалифатического изофорондиизоцианата, протекает с образованием, в основном, продукта взаимодействия изоцианатных групп c блокирующим агентом, а изоциануратные циклы и продукты вторичных реакций содержатся в незначительном количестве. В случае БПИ на основе ТДИ за счет наличия ароматического кольца, по-видимому, происходит активация второй менее реакционноспособной группы. В результате в конечном продукте присутствуют продукты взаимодействия изоциантных групп с блокирующим агентом, изоциануратные циклы и продукты вторичных реакций. Методом дифференциальной сканирующей микрокалориметрии определены температуры начала заметного разложения полученных БПИ, лежащие в интервале 120-175оС (в зависимости от структуры диизоцианата и блокирующего агента).
ОЛИГОМЕРИЗАЦИЯ ПРОПИЛЕНА И С6-ОЛЕФИНОВ НА ГЕТЕРОПОЛИКИСЛОТАХ
Зульфугарова С.М., Абдуллаева Ф.А., Муншиева М.К.
Институт Химических Проблем Национальной Академии Наук,
Баку, Азербайджан, chem@dcacs.ab.az
В последние годы вопросы охраны окружающей среды приобрели первостепенное значение, в связи с чем предъявляются более повышенные требования к химическим процессам и катализаторам.
Наши разработки позволили предложить для олигомеризации пропилена и С6-олефинов высокоактивный низкотемпературный экологически чистый катализатор, которым являются некоторые гетерополикислоты (ГПК) 12-го ряда.
В олигомеризации пропилена активными оказались фосфорно-вольфрамовая и кремневольфрамовая ГПК как в массе, так и нанесенные на силикагели различной дисперсности. Установлено, что образец, содержащий 15% фосфорно-вольфрамовой кислоты на силикагеле с удельной поверхностью 300 м2/г при температуре140-1800С, давлении 5 МПа и объемной скорости 4-7 ч-1 способствует 80-87% конверсии пропилена в ди-, три- и тетрамеры с пределами выкипания 70-220 0С.
Эти ГПК также проявили наибольшую активность в олигомеризации С6–олефинов. Процесс протекает при атмосферном давлении и температуре 720С с 60-70%-ной конверсией исходного сырья. Фосфорно-молибденовая ГПК значительно менее активна, следовательно ряд кислотности ГПК 12-го ряда H3PW12O40 ³ H4SiW12O40 > H3PMo12O40 совпадает с рядом активности этих кислот в процессе олигомеризации олефинов.
Установлено, что при нанесении ГПК на силикагель количество центров, определенное по теплотам адсорбции аммиака, соответствует числу протонов в кислоте, однако их распределение по силе отличается от такового в массивной ГПК. Из общего числа кислотных центров лишь 20% обладают такой же силой, как и в массивной ГПК, остальные же становятся значительно более слабыми. С учетом этого факта, а также того, что конверсия олефинов на нанесенных на силохром ГПК почти соответствует конверсии на соответствующих массивных ГПК, можно сделать вывод, что как на массивных, так и на нанесенных ГПК процесс олигомеризации протекает на внешней поверхности катализатора.
ПРОЦЕСС ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ КАРБОНАТОВ НА ОСНОВЕ ГЛИЦИДИЛОВЫХ ЭФИРОВ КИСЛОТ ФОСФОРА
Ильичёва О.М., Зиязетдинова В.Р., Андрианова К.А., Амирова Л.М.
Казанский государственный технический университет им. А.Н.Туполева
Казань, Россия, E-mail: molodspec@mail.ru
Циклокарбонатные связующие в последнее время привлекают все большее внимание исследователей. Высокая реакционная способность циклокарбонатов и доступность исходного сырья делают перспективным широкое применение этих продуктов в промышленности. Циклокарбонаты используются в качестве растворителей полимеров, экстрагентов ароматических углеводородов в синтезе лекарственных препаратов и полимерных материалов.
Целью данной работы явилось изучение процесса получения циклических карбонатов на основе глицидиловых эфиров кислот фосфора (ГЭФ).
Реакцию получения циклокарбонатных эфиров кислот фосфора проводили по стандартной методике, применяемой к эпоксидным олигомерам. Структуру полученных соединений изучали с помощью ИК-спектроскопии и ЯМР. На ИК спектрах обнаружено уменьшение интенсивности колебаний в области 830-840 см-1, 920 см-1, характерных для эпоксидных групп и появление частот поглощения, характерных для карбонатных групп в области 1750-1800 см-1.
В зависимости от мономера, типа инициатора или катализатора реакция может протекать по катионному, анионному или координационному механизму. В данной работе в качестве катализаторов использовали соли редкоземельных элементов (РЗЭ). Эти катализаторы стабильны и не теряют свою активность даже в водном растворе.
Выявлено, что в ходе реакции одновременно идут три процесса: образование циклокарбонатов, полимеризация ГЭФ с участием диоксида углерода и гомополимеризация глицидиловых эфиров кислот фосфора без СО2. Процессы протекают одновременно и «конкурируют» между собой в зависимости от условий синтеза (температура, давление, концентрация СО2 и др.). Исследована роль активности катализатора, показано, что с ее увеличением ускоряется процесс полимеризации и уменьшается выход циклокарбонатного продукта. При использовании низкоактивных солей РЗЭ, наоборот, наблюдается замедление процесса полимеризации и рост циклического продукта.
СИНТЕЗ ОЛИГОЭФИРФОСФОРНЫХ КИСЛОТ И ИХ ПРИМЕНЕНИЕ ДЛЯ САМООТВЕРЖДАЮЩИХСЯ ЭЛЕКТРОПРОВОДНЫХ КОМПОЗИТОВ ПОНИЖЕННОЙ ГОРЮЧЕСТИ
Константинова Е.П., Николаев П.В.
Ивановский государственный химико-технологический университет г.Иваново, Россия, e-mail: Nikolaev-prof-epoxy@yandex.ru
Для получения электропроводных эпоксидных связующих пониженной горючести нами разработана технология получения олигоэфирфосфорных кислот (ОЭФК) – производных эпоксидных олигомеров (ЭО), ортофосфорной кислоты (ОФК) и пентоксида фосфора. За счет протекания реакции гидроксифосфорилирования (раскрытия a-оксидного цикла гидроксильными группами кислоты) получены линейные ОЭФК структуры Ф1 – (ОЭ – Ф1)n (1) , содержащие фрагменты ОФК в основной цепи, где Ф1 – фрагмент ОФК, ОЭ – фрагмент олигоэфира-ацильного производного ЭО. Продукты олигомераналогичных превращений структуры (1) с фрагментами ОФК в боковой цепи Ф1–[ОЭ-(Ф2)m-Ф1]n структура (2) получены за счет реакции фосфорилирования – взаимодействия пентоксида фосфора с гидроксильными группами, которые образовались при раскрытии a-оксидного цикла кислотой и содержались во внутрицепном диарилглицериновом фрагменте дианового ЭО. Применение ОФК и пентоксида фосфора в реакциях с функциональными группами ЭО позволяет ввести в структуру получаемых фосфорилированных производных один из наиболее перспективных антипирирующих элементов - фосфор. Благодаря способности ОФК реагировать с ЭО по всем трем гидроксильным группам, были получены ОЭФК не только линейной, но и сетчатой структуры.
Показана способность некоторых ОЭФК к химическому самоотверждению. На их основе получены эпоксидные композиты (ЭК), наполненные электропроводными дисперсными наполнителями (ЭДН), и испытаны в качестве конопаточных порошков для крепления медного провода в тело электрощеток. Наилучшие результаты показали порошковые ОЭФК в смесях с ЭДН – медью ПМС-1. В результате применения такого ЭК переходное электрическое сопротивление снижается до значений 0,6-0,7 мОм (норма: не более 1,25), усилие вырывания провода достигает 25-30 кг (норма: не менее 12 кг). Во всех случаях адгезия отвержденного ЭК к проводу и щетке хорошая.
олигомерные металлокоМПЛЕКСЫ
ТЕТРА-(2,3-ПИРИДИНО)ПОРФИРАЗИНА
Корсакова И.Е., Корженевский А.Б., Койфман О.И.
Ивановский государственный химико-технологический университет,
Институт химии растворов РАН, Иваново, Россия, kobuko@isuct.ru
Уникальные свойства олигомерных металлофталоцианинов с непрерывным p-электронным сопряжением по всей макромолекуле вызвали интерес к их азаизологам.
Введение атомов азота, обладающих неподелёнными электронными парами, на периферию формального мономера, т.е. переход от фталоцианинового элементарного звена, например к тетра-(2,3-пиридино)порфиразиновому, позволяет создать в одной макромолекуле сочетание порфиразиновых и азакраун макроциклов. Такие гибридные структуры с различными геометрическими и электронными параметрами координационно-активных центров являются основой создания новых высокоэффективных материалов, обладающих каталитическими, координационными, полупроводниковыми свойствами.
Металлокомплексы олигомерного сетчатого тетра-(2,3-пиридино)порфиразина (I) синтезированы посредством темплатной полициклотетрамеризации 2,3,5,6-тетрацианпиридина в присутствие солей Cu2+, Co2+ или Fe2+ и каталитического количества молибдата аммония.
П
 олученные соединения представляют собой олигомерные структуры, вне зависимости от природы используемого металла степень полимеризации полимерного лиганда не превышает шести.
олученные соединения представляют собой олигомерные структуры, вне зависимости от природы используемого металла степень полимеризации полимерного лиганда не превышает шести. I
Идентификация структуры полимеров проведена по электронным спектрам поглощения, данным элементного анализа, а также величине молекулярной массы, приходящейся на одну концевую группу.
С
I
оединения (I) – мелкокристаллические, неплавкие до 3000С, гигроскопичные, не растворимые в воде и большинстве органических растворителей порошки черного цвета с металлическим блеском. За счёт наличия в их структуре концевых групп, а также большого количества центров протонирования они растворяются в ДМСО, ДМФА и концентрированной Н2SO4 с образованием ярко окрашенных I.
СИНТЕЗ И ИССЛЕДОВАНИЕ ПОЛИХЕЛАТОВ НА ОСНОВЕ ДИГИДРАЗИДА 2,6-ДИМЕТИЛ-3,5-ПИРИДИНДИКАРБОНОВОЙ КИСЛОТЫ И ИОНОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ
Косицина Е.С., Пугачева А.С., Нестерова Е.Ю.
Днепропетровский национальный университет,
г. Днепропетровск, Украина, len@nesterov.in.dp.ua
И
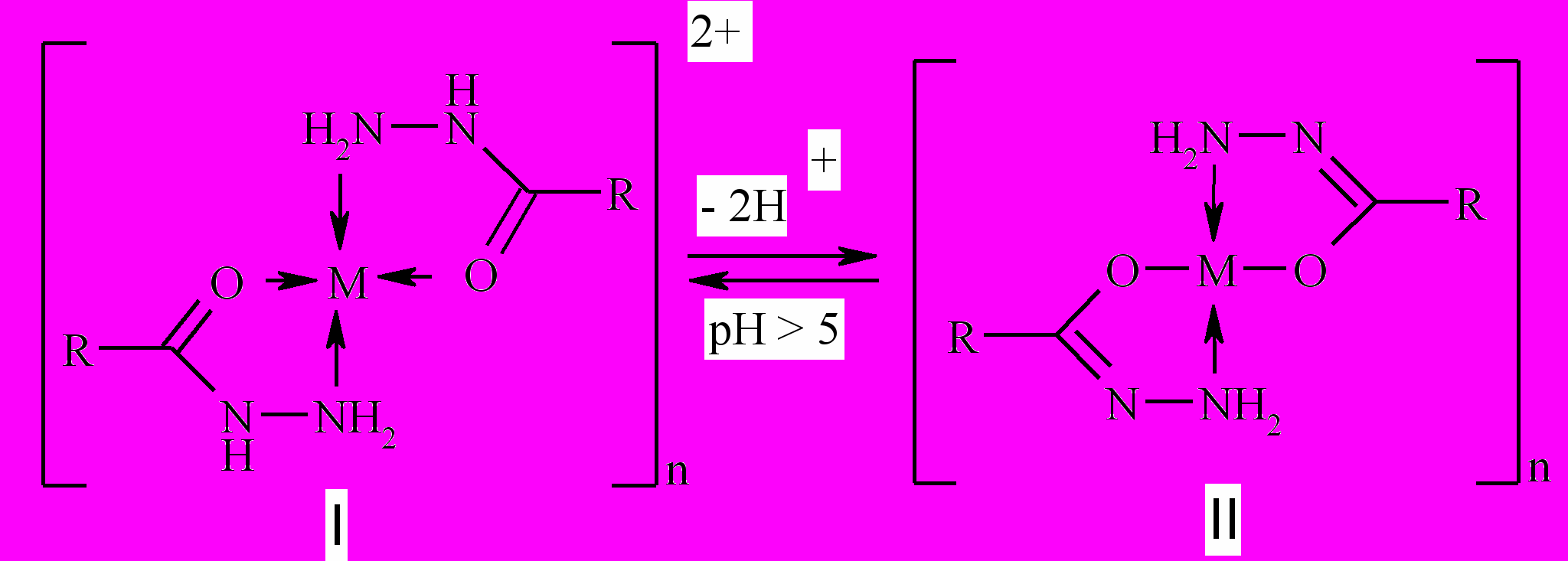 звестно, что гидразиды ароматических и гетероциклических кислот, в том числе изоникотиновой кислоты, образуют комплексные соли, строение которых, в зависимости от рН среды образования представлено в виде:
звестно, что гидразиды ароматических и гетероциклических кислот, в том числе изоникотиновой кислоты, образуют комплексные соли, строение которых, в зависимости от рН среды образования представлено в виде:В тоже время свойства полихелатов на основе дигидразидов карбоновых кислот и ионов переходных металлов изучены крайне слабо.
Нами были получены полихелаты на основе водорастворимого дигидразида 2,6-диметил-3,5-пиридиндикарбоновой кислоты и водных растворов солей Cu(NO3)2, CuSO4 и Co(NO3)2, CoSO4. С помощью УФ-спектроскопии показано, что образование полихелатов происходит в водных растворах при рН 7 в течение некоторого времени, о чем свидетельствует нарастание оптической плотности растворов при длительном выдерживании. Ход кривых насыщения, а также данные, полученные с помощью метода изомолярных серий свидетельствуют о том, что в нейтральной среде полихелаты образуются при соотношении лиганд : металл = 1:1. Это соотношение подтверждается также данными элементного анализа выделенных продуктов.
В ИК-спектрах полученных полихелатов наблюдается низкочастотный сдвиг на 80-150 см-1 полосы поглощения n(NH2) и на 10-15см-1n(С=О) по сравнению с аналогичными полосами свободного лиганда, что свидетельствует о предпочтительной реализации комлексной формы I. Изучена термическая и химическая устойчивость полученных полихелатов методами ДТА и ДГА. Найдено, что термическая устойчивость полихелатов сильно зависит от природы аниона и для сульфатов существенно выше (Тразл. = 400-5000С), чем для нитратов (Тразл. = 200-3000С). На основании полученных данных предложена структура внутренней сферы полученных полихелатов с учетом природы координационного металла и соответствующего аниона.
КИНЕТИЧЕСКИЕ ЗАКОНОМЕРНОСТИ ОБРАЗОВАНИЯ ОЛИГОМЕРНЫХ ПРОДУКТОВ ОКИСЛЕНИЯ БИЦИКЛООЛЕФИНОВ НОРБОРНЕНОВОГО РЯДА
Куликова Т.А., Мачтин В.А., Плисс Е.М.1
Ярославский государственный технический университет,
1Ярославский государственный университет им. П.Г. Демидова,
г. Ярославль, Россия, pliss@uniyar.ac.ru
Исследования окисления бициклоолефинов норборненового ряда, проведенные в 80-х годах прошлого века, привели авторов (Э.А. Блюмберг с сотр.; В.В. Вороненков с сотр.) к выводу, что реакция продолжения цепи протекает как стереоспецифическое цис-экзо-присоединение пероксирадикала к p-связи бициклоолефина в соответствии с нижеприведенной схемой:
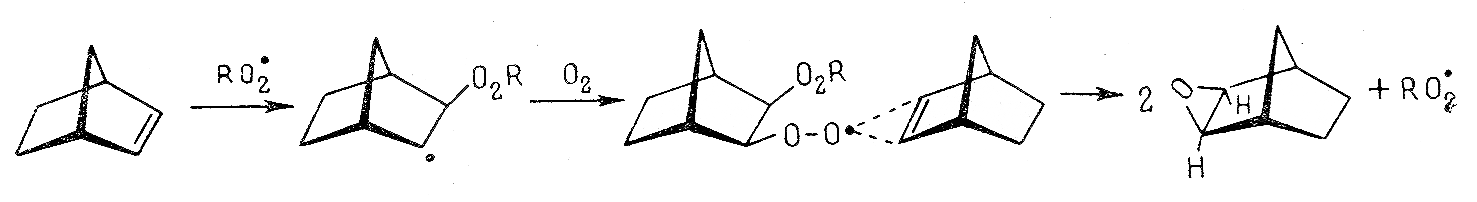
Нами установлено, что эпоксид – лишь один из продуктов окислительных превращений таких соединений. Для этого использована комбинация методов микроволюмометрии, ЭПР, ИК-Фурье-спектроскопии, классического ЯМР и Н-Н двумерной NOESY-спектроскопии, жидкостной и гель-хроматографии и полярографии в сочетании с компьютерным моделированием и квантово-химическими расчетами. Показано, что при окислении норборнена, винилнорборнена, этилиденнорборнена и норборнодиена образуются как низкомолекулярные (гидропероксиды и эпоксиды), так и олигомерные продукты (пероксиды).
Методами микроволюмометрии и ЭПР показано, что фенолы и ароматические амины тормозят накопление как низкомолекулярных, так и олигомерных продуктов окисления, в то время как стабильные нитроксильные радикалы замедляют образование только низкомолекулярных соединений.
Приведенные факты обусловлены феноменологией процесса окисления бициклоолефинов, связанной с тем, что пероксирадикалы атакуют различные реакционные центры (p- и CH-связи). Причем при атаке p-связей образуются олигомерные пероксиды и эпоксиды, а при взаимодействии пероксирадикалов с CH-связями - низкомолекулярные гидропероксиды.
НОВЫЙ СПОСОБ СИНТЕЗА ГИПЕРРАЗВЕТВЛЕННЫХ ПОЛИМЕРОВ МЕТОДОМ ТРЕХМЕРНОЙ РАДИКАЛЬНОЙ ПОЛИМЕРИЗАЦИИ ВИНИЛОВЫХ МОНОМЕРОВ В ПРИСУТСТВИИ КИСЛОРОДА
Курочкин С.А., Грачев В.П., Королев Г.В.