
Секция 1 Аналитическая химия
| Вид материала | Доклад |
- Рабочая программа дисциплины (модуля) «Линейная алгебра и аналитическая геометрия», 275.82kb.
- Рабочая программа по дисциплине «Спектральные методы анализа» для специальности 020101, 175.88kb.
- Рабочая программа дисциплины (модуля) «математический анализ», 424.74kb.
- Рабочая программа дисциплины (модуля) «Уравнения математической физики», 266.58kb.
- Рабочая программа дисциплины аналитическая химия Направление подготовки, 1181.86kb.
- Неорганическая и аналитическая химия, 221.14kb.
- Программа «аналитическая химия» по направлению подготовки 020100 «Химия», 31.74kb.
- Рабочей программы учебной дисциплины аналитическая химия уровень основной образовательной, 52.53kb.
- Примерная программа наименование дисциплины «Неорганическая и аналитическая химия», 341.23kb.
- Конспект лекций по курсу «Неорганическая и аналитическая химия», 18.21kb.
Изучение влияния посторонних ионов и маскирующих веществ на комплексообразования германия(IV) в виде бинарного и разнолигандных комплексов показало, что в присутствии поверхностно-активных веществ значительно увеличивается избирательность реакции. Установлено, что разработанные методики определения германия(IV) с реагентом в присутствии цетилтриметиламмоний бромистого, цетилпиридиний бромистого и цетилпиридиний хлористого обладают высокой избирательностью. Разработана высокоселективная методика фотометрического определения германия(IV) в полупроводниковом материале.
ОПРЕДЕЛЕНИЕ АРОМАТИЧЕСКИХ РАСТВОРИТЕЛЕЙ В ЧЕРНИЛАХ ШАРИКОВЫХ РУЧЕК МЕТОДОМ ВЭЖХ С ФЛУОРИМЕТРИЧЕСКИМ ДЕТЕКТИРОВАНИЕМ
Шевченко Т.Н.
Кубанский государственный университет,
Краснодар, Россия.
Аспирант 3г.
tyler22286@mail.ru
Научный руководитель: Темердашев З.А.
Для установления давности выполнения записей при проведении судебно-технической экспертизы документов одним из критериев является контроль содержания ароматических спиртов, которые используются в качестве консервантов и растворителей чернил. Существующие методики их определения основаны на изучении динамики испарения растворителей из штриха во времени с использованием газовой хроматографии/масс-спектрометрии.
Нами показана возможность применения ВЭЖХ с флуориметрическим детектированием в качестве экспрессной, селективной и чувствительной альтернативы для определения ароматических спиртов – бензилового спирта и 2-феноксиэтанола – основных и наиболее распространённых растворителей в составе чернил шариковых ручек. Экспериментальные исследования показали, что для этих целей наиболее оптимален обращено-фазовый вариант хроматографии с градиентным элюированием. Флуориметрическое детектирование выбрано в качестве наиболее чувствительного метода, практически не уступающего по чувствительности ГХ-МС.
В качестве объектов испытаний анализировались записи, выполненные чернилами шариковых ручек синего, фиолетового и черного цветов различных стран и фирм-производителей (Китай, Индия, Турция, Корея, Япония, Россия, Италия, Франция, Германия, Австрия, США).
Изучены факторы, влияющие на условия скрининга и детектирования анализируемых образцов. Во всех испытуемых образцах в качестве преобладающего растворителя определялся 2-феноксиэтанол, а содержание бензилового спирта в различных чернилах варьировалось от единиц процентов до следовых количеств. Показана возможность проведения судебно-технических экспертиз документов по результатам идентификации и оценки содержания растворителей, которые могут выступать маркерами «старения» чернил шариковых ручек.
СОЗДАНИЕ ИССЛЕДОВАНИЕ ХАРАКТЕРИСТИК ИОНСЕЛЕКТИВНОГО ЭЛЕКТРОДА С ОТКЛИКОМ НА ДИАЗОЛИН
Шевчук И.К.
Тверской государственный университет,
Тверь, Россия.
Студент III курса.
irena-shevchuk@mail.ru
Научный руководитель: Мантров Г.И.
Потенциометрические методы, в частности, с использованием ионоселективных электродов (ИСЭ) выгодно отличаются простотой и экспрессностью анализа, однако применение этого метода для определения диазолина не описано в литературе, поэтому целью настоящей работы явилось создание ИСЭ для определения последнего, изучение его характеристик и разработка методики ионометрического определения диазолина в готовых лекарственных формах.
В работе использовали диазолин фармакопейной чистоты, фосфорно-молибденовую (ФМК) и фосфорновольфрамовую (ФВК) кислоты ч.д.а., диоктилфталат (ДОФ) ч.д.а., поливинилхлорид (ПВХ) марки С-70 х.ч. Электродноактивные вещества (ЭАВ) получали осаждением диазолина из водных растворов вышеуказанными гетерополикислотами.
Пластифицированные мембраны имели следующий состав (в масс. %): ПВХ-35, ДОФ-60, ЭАВ-5. ИСЭ перед применением вымачивали в 0,05 М растворе диазолина. Для определения электродных характеристик использовали электрохимическую ячейку:
| Ag/AgCl | 2.08 * 10 –3 M р-р а/б + 0.1 М р-р KCL | Пленочная мембрана | Исследуемый раствор | AgCl | Ag |
Изготовленные электроды обладали хорошими метрологическими характеристиками. Интервал линейности электродной функции находится в промежутке 1-5 рС, крутизна электродной функции близка к теоретическому значению, время отклика составляло 5-10 с. Показано, что потенциал ИСЭ не изменяется в интервале рН 4 - 8, что делает этот интервал наиболее подходящим для определения диазолина. Определение диазолина в готовых лекарственных формах показало работоспособность созданного ИСЭ.
Литература:
[1] Rakhmanko E. M., Yegorov V. V., A. L. Gilevich A. L. Ion-Sel. Electrode, 5-11 (1992)
[2] Корыта И., Штулик К. Ионоселективные электроды. - М.: Мир, 1989, 268 с.
СОДЕРЖАНИЕ И ИЗОТОПНЫЙ СОСТАВ УРАНА В МИНЕРАЛЬНОЙ ВОДЕ "ЕССЕНТУКИ № 17"
Ширяева Ю.В.
Санкт-Петербургский государственный университет,
Санкт-Петербург, Россия.
Студент V курса.
gwettarrin@rambler.ru
Научный руководитель: Пучкова Е.В.
Различное химическое поведение материнского (238U) и дочернего (234U) изотопов при инконгруентном растворении урансодержащих горных пород природными водами приводит к обогащению вод минеральных источников радиогенным изотопом 234U. Например, известно [1], что отношение активностей изотопов
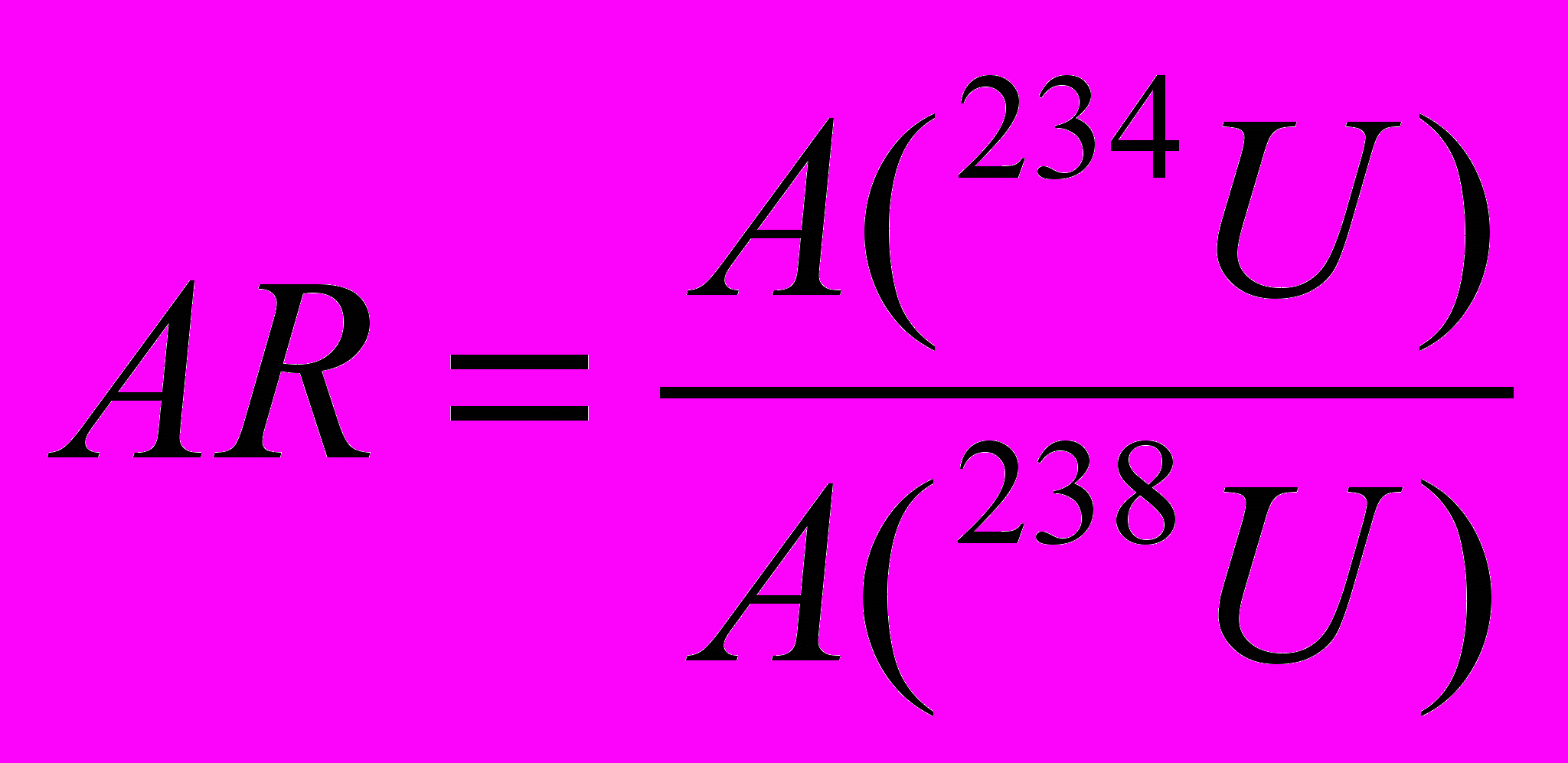 (Бк/Бк) в минеральной воде «Нарзан» составляет 1,4 – 1,6. Для воды «Джермук» это отношение находится в пределах 1,51 – 1,68, для источника «Арзни» повышается до 1,85-2,44. Что касается минеральных вод «Ессентуки», то, несмотря на более чем полуторавековую историю их использования, в литературе практически отсутствуют данные о величинах AR.
(Бк/Бк) в минеральной воде «Нарзан» составляет 1,4 – 1,6. Для воды «Джермук» это отношение находится в пределах 1,51 – 1,68, для источника «Арзни» повышается до 1,85-2,44. Что касается минеральных вод «Ессентуки», то, несмотря на более чем полуторавековую историю их использования, в литературе практически отсутствуют данные о величинах AR. Целью представляемой работы являлось исследование содержания и изотопных характеристик урана в минеральной воде «Ессентуки № 17», отобранной из скважины № 47 Нагутского месторождения.
Исследованию подвергались образцы минеральной воды различных производителей, представленных в торговой сети Санкт-Петербурга. Выбор данной марки обусловлен наличием наибольшего числа производителей, достигающего, по разным сведениям, 13 и даже 27. Информация об исследованных образцах представлена в таблице.
Таблица Сведения об исследованных образцах минеральной воды «Ессентуки № 17»
| Шифр образца | Производитель | Минерализация, г/л | Концентрация 238U, мкг/л | AR | Примечание |
| 1а | ООО Универсальный завод розлива минеральной воды «АКВА-ВАЙТ» | 10 | 0,040,01 | 1,120,08 | Дата розлива 15.08.2011 г. |
| 1б | 0,240,05 | 1,020,03 | Дата розлива 20.10. - 11.11.2011 г. | ||
| 2 | Vita | - | < МДА | - | |
| 3 | ООО «Ессентукский завод минеральн ых вод на КМВ» | 10,9 | < МДА | - | |
| 4 | ООО «Кавказская здравница» | 12,5 | 0,080,02 | 1,050,14 | |
| 5 | ЗАО «КМВ-Пластик» | 10,8 и 11,9 | 0,100,02 | 1,190,17 | |
| 6 | ООО «Элита-Минерал групп» | 7 и 8,9 | 0,420,08 | 1,130,09 | |
Для выделения урана была разработана методика, включающая основные принципы радиохимического анализа объектов естественного происхождения. Пробу воды объемом 6 л подкисляли концентрированной азотной кислотой, вводили изотопную метку 232U (для определения химического выхода) и кипятили в течение 10-15 минут для разрушения карбонатов. Изотопы урана концентрировали на неспецифическом носителе Fe(OH)3, который осаждали безугольным аммиаком. Выделение урана выполняли методами радиохимического анализа. Концентрацию 238U и величину AR определяли методом альфа-спектрометрии. Химические выходы составляли 30-60%. Для определения минерализации воды 1 л пробы упаривали до сухого солевого остатка.
Обнаружено, что показатели минерализации большинства проб соответствуют диапазону значений, заявленному производителями. Исключением являются образцы производителя № 6. Масса солевого остатка в одном образце составила 7 г/л, в другом – 8,9 г/л при заявленном диапазоне значений 9,2 – 13,0 г/л.
Значения AR в образцах лежат в интервале 1.00 – 1.09, что незначимо отличается от равновесной величины.
Концентрации 238U варьируют в очень широком диапазоне: от фоновых значений в образцах № 2 и № 3 до 0,42 мкг/л в образце № 6. При этом следует отметить, что максимальная концентрация урана обнаружена в образце с наименьшей минерализацией. В образцах № 1а и № 1б концентрации 238U различаются в 6 раз, несмотря на то, что единственным различием образцов является дата розлива из скважины.
Таким образом, при одинаковом химическом составе минеральной воды «Ессентуки № 17», заявленном производителями, содержание микрокомпонента 238U различается на порядок (образцы 1а и 6). Этот факт позволяет сделать вывод о том, что минерализация ряда образцов имеет не природное происхождение.
Литература:
[1] Чалов, П.И. Изотопное фракционирование природного урана. − Фрунзе: Илим, 1975, 150 c.
ИДЕНТИФИКАЦИЯ ПРИМЕСЕЙ ОРГАНИЧЕСКИХ КИСЛОТ В ЯНТАРНОЙ КИСЛОТЕ МЕТОДОМ ВЭЖХ
Шумилин А.С.
Тульский государственный педагогический университет им. Л.Н. Толстого,
Тула, Россия.
Молодой учёный.
shumilin-as@mail.ru
Научный руководитель: Шахкельдян И.В.
В медицине, фармацевтике и ветеринарии широкую известность получили лекарственные препараты на основе янтарной кислоты, обладающей широким спектром воздействия на различные механизмы регуляции метаболической активности клеток и тканей, находящихся в состоянии возбуждения или патологически измененных. Безвредность янтарной кислоты и ее соединений и способность оказывать положительный эффект при низких дозировках делают возможность ее использования при разработке нового поколения лекарств [1]. Янтарная кислота обладает высокой адаптогенной, антигипоксической, антиоксидантной, нейротропной активностью, оказывает выраженное нормализующее действие на энергетический обмен и процессы биосинтеза в условиях патологий и экстремальных состояний [2, 3].
В последнее время ведется множество разработок по биотехнологическому способу получения многих ценных продуктов, в том числе ФК и ЯК [4], однако селективность микробиологических способов не столь высока, для того чтобы получать продукт соответствующей степени чистоты, предъявляемой к фармпрепаратам. Кроме того, выделение и очистка продукта требует дополнительных затрат и оборудования. В качестве примесей могут присутствовать другие органические кислоты (лимонная, щавелевая, яблочная, глутаровая и др.), которые сложно разделить физическими способами. Наличие примесей снижает качество янтарной кислоты как химического реактива и фармацевтического препарата. Получение янтарной кислоты гидрированием малеинового ангидрида на палладиевых катализаторах [5] априори обеспечивает отсутствие выше перечисленных примесей.
Для установления состава примесей или их отсутствия мы использовали метод высокоэффективной жидкостной хроматографии (ВЭЖХ). В образцах янтарной кислоты, полученные гидрированием малеинового ангидрида необнаружено ни примесей непредельных кислот (фумаровой и малеиновой), которые могли бы образовываться из субстрата, а также продуктов более глубокого восстановления (гидрирование карбоксильных групп). В образцах янтарной кислоты полученных как отходы промышленного производства адипиновой кислоты были обнаружены пики соответствующие глутаровой и адипиновой кислот, что не позволяет использовать кислоту такого качества в фармацевтике. Образцы, полученные биотехнологическими способами имеют следующий набор примесей: кетоглутаровая, лимонная, щавелевая кислоты и ряд других неидентифицированнных примесей.
Таким образом, установлено что гидрирование малеинового ангидрида в водной среде на палладиевых катализаторах ведет к получению янтарной кислоты высокого качества, в отличие от окислительных и биотехнологических способов синтеза.
Литература:
[1] Янтарная кислота в медицине, пищевой промышленности, сельском хозяйстве: сборник научных статей/ Ин-т теор. и экспер. биофизики РАН; под ред. проф. М.Н.Кондрашовой. Пущино: ИТЭБ, 1996.
[2] Коваленко А.Л., Белякова Н.В. Фармация 5 (2000)
[3] Лебедев А.Ф., Швец О.М., Евглевская Е.П., Евглевский А.А. Научно-практические аспекты разработки новых иммунометабо-лических препаратов на основе янтарной кислоты, их клиническая и производственная эффективность [Янтарный биостимулятор, металлосукцинат и металлосукцинат плюс] монография. - Курск: Изд-во Курской гос. с.-х. акад., 2010, 113 с.
[4] Юсупова А.И. Биосинтез янтарной кислоты дрожжами из ятанола// Автореферат дисс. Канд. биол. Наук. – Пущино, 2011
[5] Кондрашова М.Н., Любимова Т.Б., Маевский Е.И., Пивоненкова Л.П., Учитель М.Л., Хейфец В.И., Чекова О.А. Способ получения янтарной кислоты. Патент РФ № 2237056., 2003.
ХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ МОДИФИЦИРОВАННЫХ ПАН ВОЛОКОН
Щербина Н.А.
Балаковский институт техники, технологии и управления (филиал) Саратовского государственного технического университета,
Балаково, Россия.
Молодой учёный.
nat80371@yandex.ru
Научный руководитель: Панова Л.Г.
Для снижения горючести ПАН сополимера необходимо инициировать процессы циклизации, обеспечивая снижение выхода летучих горючих продуктов в газах пиролиза.
В качестве замедлителей горения использовались полифункциональные органические соединения метилфосфонамид и N-метилол-3-(диметилфосфинил)пропионамид, которые разлагаются с эндотермическим эффектом в температурном интервале основной стадии деструкции ПАН волокна.
Изучение состава продуктов пиролиза проводили с использованием хроматографа–масс-спектрометра Focus GC/DSG. Пиролиз проводили в среде гелия при 750-11000С. Колонка капиллярная DB-5 диаметром 0,25 мм, длина 15 метров. Масса навески – 50 мг. Режим хроматографирования: температура термостата 340С, скорость нагрева 350С/мин. до 3000С, температура инжектора 2500С, поток газа 1мл/мин., делитель потока 1:50.
Подтверждается влияние ЗГ на процессы пиролиза. Анализ хроматограмм исходного и модифицированного волокна показал больший выход газообразных продуктов у модифицированного волокна в первые 90-120с.
Таблица 1. ГХ-МС- анализ ПАН волокон при пиролизе
| Брутто формула | Название основных выделяющихся продуктов, N°-CAS | Исходное ПАН волокно | Модифицированное ПАН волокно | ||
| RT, мин. | m/е (%) | RT, мин. | m/е (%) | ||
| C3H3N | акрилонитрил CAS:107-13-1 | 1,29 | 26(98) 53(100) | 1,70 | 53(100) 26(10) |
| C2H3N | ацетонитрил CAS:75-05-8 | 1.62 | 41(100) | - | - |
| C6H6 | бензол CAS:71-43-2 | 2,46 | 78(100) | 2,45 | 78(100); 51(25); 39(14) |
| C7H8 | толуол CAS:108-88-3 | 3,20 | 91(100) | 3,20 | 91(100); 92(70) |
| C8H8 | стирол CAS:100-42-5 | 3,98 | 104(100) | 3,98 | 104(100); 78(34); 51(25) |
| C7H5N | бензонитрил | 4,38 | 103(100) | 4,39 | 103 |
| C8H7N | 4метилбензо нитрил CAS:104-85-8 | 4,48 | 117(100) | 4,91 | 117(100); 90(25); 63(12); 51(8); 39(15) |
| C10H8 | нафталин CAS:91-20-3 | 5,39 | 128(100) | 5,39 | 128(100); 102(7); 77(5); 64(10); 51(10) |
| C8H14N2 | 1.3-дицианобензол CAS:626-17-5 | 5,52 | 128(100) 101(18); 75(10); 64(7); 50(12) | 5,79 | 128(100) |
| C11H7N | 1-цианонафталин CAS:1984-04-9 | 6,51 | 126(27); 153(100) | 6,51 | 153(100) |
| C11H7N | 2-цианонафталин CAS:613-46-7 | 6,59 | 153(100) 126(13) | - | - |
| C12H8 | аценафталин CAS:208-96-8 | 6,40 | 152(100) | - | - |
| C14H10 | антрацен CAS:120-12-7 | 7,48 7,33 | 178(100) 177(88) | 7,48 - | 178(80); 177(100); 152(15); 151(18); 89(18); 76(25) |
| C16H12 | 1-фенилнафталин CAS:605-02-7 | 8,43 | 202(100) | - | - |
Продукты разложения ЗГ инициируют формирование КО. При повышении температуры от 7500С до 1100 0С. уменьшается образование газообразных продуктов у модифицированного ПАН волокна.