
Секция 1 Аналитическая химия
| Вид материала | Доклад |
- Рабочая программа дисциплины (модуля) «Линейная алгебра и аналитическая геометрия», 275.82kb.
- Рабочая программа по дисциплине «Спектральные методы анализа» для специальности 020101, 175.88kb.
- Рабочая программа дисциплины (модуля) «математический анализ», 424.74kb.
- Рабочая программа дисциплины (модуля) «Уравнения математической физики», 266.58kb.
- Рабочая программа дисциплины аналитическая химия Направление подготовки, 1181.86kb.
- Неорганическая и аналитическая химия, 221.14kb.
- Программа «аналитическая химия» по направлению подготовки 020100 «Химия», 31.74kb.
- Рабочей программы учебной дисциплины аналитическая химия уровень основной образовательной, 52.53kb.
- Примерная программа наименование дисциплины «Неорганическая и аналитическая химия», 341.23kb.
- Конспект лекций по курсу «Неорганическая и аналитическая химия», 18.21kb.
РАЗРАБОТКА И ВАЛИДАЦИЯ СПЕКТРОФОТОМЕТРИЧЕСКИХ МЕТОДИК ОПРЕДЕЛЕНИЯ СУЛЬФАМЕТАЗИНА В МНОГОКОМПОНЕНТНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВАХ С ИСПОЛЬЗОВАНИЕМ АЗОРЕАГЕНТОВ: ТРОПЕОЛИНА О, 4-(2-ПИРИДИЛАЗО) РЕЗОРЦИНА И 4-(2-ТИАЗОЛИЛАЗО) РЕЗОРЦИНА
Коркуна О.Я.,1 Бойко М.Я.2
1Львовский национальный университет имени Ивана Франко,
Львов, Украина.
Молодой учёный.
olga_korkuna@yahoo.com
2Львовский национальный университет имени Ивана Франко, Львов, Украина. Аспирант 3г.
Научный руководитель: Врублевская Т.Я.
Сульфаниламиды (СА) являются одним из старейших классов антибактериальных средств. На фоне сокращения использования СА, как однокомпонентных препаратов, широкое применение получили комбинированные препараты, содержащие СА в сочетании с триметопримом, либо с некоторыми антибиотиками. Такие комбинации антибактериальных субстанций характеризуются выраженным бактерицидным эффектом и широким спектром антибактериальной активности, в т. ч. относительно микрофлоры, устойчивой ко многим антибиотикам и издавна используемым сульфаниламидам.
Известно, что как действующие, так и вспомогательные вещества, составляющие один комбинированный препарат, очень часто затрудняют определение каждого, либо некоторых, из них. Этим, в частности, обуславливается необходимость поиска селективных реагентов для определения, контролируемых веществ, как среди уже известных соединений, так и поиск новых аналитических форм.
Наиболее распространенным СА, используемым в ветеринарной практике является сульфаметазин (4-Амино-N-(2,4-диметил-2-пиридинил) бензосульфонамид), резорбтивный препарат краткосрочного действия. Нами изучено взаимодействие сульфаметазина (СМТ) с азореагентом (кислотным моноазокрасителем) – тропеолином О (Тр О), и гетероциклическими азореагентами 4-(2-пиридилазо) резорцином (ПАР) и 4-(2-тиазолилазо) резорцином (ТАР). Методики основаны на диазотировании СМТ 10-кратным избытком нитрита натрия в среде 1 М соляной кислоты при температуре ледяной бани в течение 20 минут. Последующее азосочетание, образовавшихся диазосолей СА, с азокрасителями Тр О, ПАР и ТАР осуществляется, соответственно, при рН 10,5, 11,0 и 9,5 при 1,5-2-кратном избитке азореагента. Для всех систем в качестве буферной смеси использовали 0,01 М раствор тетрабората натрия.
На стадии разработки методик было установлено, что вспомогательные вещества, входящие в состав лекарств, не влияют на определение СМТ с использованием всех испытанных азокрасителей, в то время, как антибиотики тетрациклинового ряда и тилозин мешают такому определению с участием ПАР и ТАР. Полученные результаты позволили применить разработанные методики для анализа лекарственных средств в различных лекарственных формах (растворы, суспензии, порошки, таблетки), с использованием, в частности, Тр О – для четырехкомпонентных, ПАР – трехкомпонентных и ТАР – двухкомпонентных препаратов.
Согласно требованиям Европейской [1], Американской [2] Фармакопей, Государственной Фармакопеи Украины [3] а также других международных [4] и внутригосударственных документов [5], методики определения действующих веществ в лекарственных средствах должны быть валидированы. В соответствии с требованиями [1-5] для аналитической методики на испытание "количественное определение содержания действующего вещества" необходимо определять такие валидационные характеристики: специфичность; робастность (стойкость); линейность; правильность (или точность), прецизионность на трех уровнях – сходимость (повторяемость), внутрилабораторную (промежуточную) прецизионность, воспроизводимость; диапазон применения; предел обнаружения и предел количественного определения.
Исходя из этих требований, мы провели валидацию разработанных нами методик: методику определения сульфаметазина с Тр О в таблетках "Септовет" (ООО Укрзооветпромпостач) и порошке "Сульфатилозин" (ООО Укрветпромпостач); с ПАР – в суспензии "Мастисан-А" (Харьковская биофабрика); с ТАР – в растворе "Веттримеразин" (ООО Базальт) (табл. 1).
Таблица 1. Состав исследуемых препаратов сульфаметазина.
| Препарат | Другие действующие вещества | Вспомогательные вещества |
| "Веттримеразин" (186±19 мг/мл) | триметоприм (40 мг/г) | пропиленгликоль, бензиловый спирт, вода |
| "Мастисан-А" (70±7 мг/мл) | стрептомицин (12 мг/мл), бензилпенницилин (26 мг/мл) | стеарат алюминия, вазелиновое масло |
| "Септовет" (180±18 мг/табл) | тилозин (25 мг/табл), триметоприм (30 мг/табл), ретинол ацетат (6 мг/табл) | лактоза, крохмаль, гидрокар бонат натрия, хлорид натрия, стеарат магния, карбоксиметил целюлоза, поливиниловый спирт, винная кислота |
| "Сульфатилозин" (175±17,5 мг/г) | триметоприм (35 мг/г), тилозин (3 мг/г), окситетрациклин (5 мг/г) | крохмаль, лактоза |
Экспериментально доказано, что спектрофотометрическая методика количественного определения СА с использованием Тр О, ПАР и ТАР в исследованных препаратах пригодна для контроля качества этих препаратов по показателю "количественное определение" СМТ, что подтверждено установленными валидационными характеристиками.
Литература:
[1] European Pharmacopoeia (Eur. Ph.). 7-th Ed. Strasbourg: Council of Europe,2010.
[2] United States Pharmacopoeia, USP 30-NF25 Convention Inc., Rockville, MD XXVI, 2007.
[3] Державна Фармакопея України. – Доп. 1. – Х.: РІГЕР, 2004. – 520 с.
[4] Note for guidance on validation of analytical procedures: text and methodology (CPMP/ICH/381/95).
[5] Юргель Н.В., Младенцев А.Л., Бурдейн А.В. и др. Руководство по валидации методик анализа лекарственных средств – М.: — Изд. «Спорт и Культура - 2000», 2007. – 192 с.
ОПРЕДЕЛЕНИЕ ОСТАТОЧНЫХ КОЛИЧЕСТВ БИСФЕНОЛА А И ДИЭТИЛСТИЛЬБЭСТРОЛА В ПРОДУКТАХ ПИТАНИЯ С ИСПОЛЬЗОВАНИЕМ СОЧЕТАНИЯ МЕТОДОВ QUECHERS И ДИСПЕРСИОННОЙ ЖИДКОСТНО – ЖИДКОСТНОЙ МИКРОЭКСТРАКЦИИ МЕТОДОМ ГАЗОВОЙ ХРОМАТОГРАФИИ
Королёв Д.С.
Федеральный центр охраны здоровья животных,
Владимир, Россия.
Аспирант 2г.
bbzanko@rambler.ru
Научный руководитель: Амелин В.Г.
Разработана простая и экономичная методика определения бисфенола А и диэтилстильбэстрола в широком круге пищевых продуктов с использованием сочетания методов QuECheRS и дисперсионной жидкостно – жидкостной микроэкстракции методом газовой хроматографии с детектором электронного захвата. Анализируемые вещества извлекали из твердых проб ацетонитрилом согласно методу QuECheRS, после чего концентрирование проводили при помощью микроэкстракции, где в качестве экстрагента был использован тетрахлорметан, диспергатора – конечный экстракт по методу QuECheRS. Микроэкстракцию проводили в среде бидистиллированной воды без добавления солей и изменения рН. Эффективность диспергирования увеличивали обработкой полученной эмульсии ультразвуком. Дериватизацию проводили трифторуксусным ангидридом при температуре 60 0С с добавлением катализатора триэтиламина.
Были подобраны оптимальные объемы и условия для микроэкстракции бисфенола А и диэтилстильбэстрола, степень извлечения которых близка к 100%, а степень концентрирования конечного экстракта - 40. Интервал линейности градуировочного графика составил 0,05 – 10 и 0,02 – 5 мкг/мл для бисфенола А и диэтилстильбэстрола соответственно. Диапазоны определяемых концентраций в мясе, креветках, мидиях, консервированных овощах и фруктах составили 1 – 250 и 0,5 – 125 мкг/кг для бисфенола А и диэтилстильбэстрола соответственно.
РАЗРАБОТКА ВАРИАНТОВ ON-LINE КОНЦЕНТРИРОВАНИЯ ПРИ ЭЛЕКТРОФОРЕТИЧЕСКОМ ОПРЕДЕЛЕНИИ БЕЛКОВ В УСЛОВИЯХ КАПИЛЛЯРНОЙ ЭЛЕКТРОХРОМАТОГРАФИИ
Королева В.Ю.
Санкт-Петербургский государственный университет,
Санкт-Петербург, Россия.
Студент IV курса.
moonrose89@mail.ru
Научный руководитель: Карцова А.А.
Капиллярная электрохроматография (КЭХ), являясь микроколоночным электрокинетическим методом разделения аналитов, обладает достоинствами ВЭЖХ и капиллярного зонного электрофореза (КЗЭ). Новые технологии изготовления колонок для КЭХ позволяют определять как высокомолекулярные соединения, так и их низкомолекулярные метаболиты. Однако, пределы обнаружения аналитов остаются достаточно высокими, что затрудняет активное использование КЭХ в практике клинической медицины. Перспективным решением является поиск соответствующих вариантов on-line концентрирования, включая комбинирование различных механизмов концентрирования, позволяющих получать сопоставимые с высокоэффективной жидкостной хроматографией (ВЭЖХ) отношения сигнал/шум.
Основными вариантами концентрирования являются стэкинг, свипинг, динамический рН-скачок, самоиндуцированный изотахофорез.
В последние годы большой интерес привлекает использование в качестве компонентов подвижной и неподвижной фаз в хроматографии и капиллярном электрофорезе новых полимерных материалов - дендритных полимеров. Они имеют стабильную мицеллоподобную структуру, большое количество терминальных функциональных групп, внутримолекулярные полости, обеспечивающие способность образовывать комплексы включения типа «гость-хозяин» с аналитами различной природы. Нами синтезированы полые колонки с нанесенным тонким слоем сверхразветвленных полимеров (PLOT-колонки). На подготовленных колонках изучены возможности различных вариантов on-line концентрирования для снижения пределов обнаружения белков (альбумина, миоглобина, лизоцима, инсулина) при их электрофоретическом определении с УФ-детектированием.
Показано, что в кислой среде с добавкой полимеров в состав буферного электролита реализуются условия стэкинга с большим объемом вводимой пробы (без переключения полярности).
Установлено, что использование стэкинга позволяет снизить пределы обнаружения белков по сравнению с традиционным капиллярным зонным электрофорезом до 70 раз.
Проведена количественная оценка on-line концентрирования и выявлены факторы, определяющие его эффективность (степень функционализации молекулы дендритного полимера; природа, концентрация и рН рабочего буферного раствора). Получены сравнительных оценочных характеристик по пределам обнаружения, эффективности, селективности разделения.
Осуществлена апробация установленных закономерностей на реальных объектах (сыворотка крови).
ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНО-МАССОВОГО РАСПРЕДЕЛЕНИЯ ОЛИГОМЕРОВ ПОЛИМЕТИЛЕННАФТАЛИНСУЛЬФОНАТА МЕТОДОМ MALDI-TOF
Краснова Т.А.
Владимирский государственный университет имени Александра Григорьевича и Николая Григорьевича Столетовых,
Владимир, Россия.
Аспирант 1г.
krasnova-ta@mail.ru
Научный руководитель: Амелин В.Г.
Моделирование свойств различных строительных материалов сейчас практически невозможно без применения специальных химических добавок.
Потребительские свойства модификаторов и их стабильность в значительной степени зависят от строения и характеристик синтезированных олигомеров и полимеров.
Существующие методы контроля, закрепленные в нормативной документации, не позволяют в полной мере оценить свойства выпускаемой продукции. А контроль потребительских свойств, помимо своей высокой трудоемкости и материалоемкости, может длиться от месяца до года.
При производстве описанных модификаторов наиболее важным параметром является молекулярно-массовое распределение. По соотношению фракций с различным числом звеньев и, соответственно, различной молекулярной массой можно определить все основные параметры качества выпускаемой продукции в считанные минуты, не затрачивая большого количества ресурсов.
Рассмотрена возможность применения масс-спектрометрии с матрично-активированной лазерной диссоциацией/ионизацией в сочетании с времяпролетным масс-анализатором (MALDI-TOF) для установления молекулярно-массового распределения олигомера полиметиленнафталинсульфоната (ПМНС) – модификатора строительных материалов на основе портландцемента.
Показано применение метода MALDI-TOF для определения молекулярно-массового распределения смеси олигомеров ПМНС в диапазоне масс от 600 Da до 6000 Da.
Использовали MALDI-TOF масс-спектрометр Autoflex III smartbeam (Bruker). Для определения использовалась протонированная форма олигомера – полиметиленнафталинсульфокислота. Для повышения разрешающей способности использовали режим работы с рефлектроном, основные параметры анализа: используется ультрафиолетовый азотный лазер с длиной волны 337 нм, с длиной импульса 5 нс и мощностью лазерного излучения в диапазоне 106 – 107 Вт/см2.
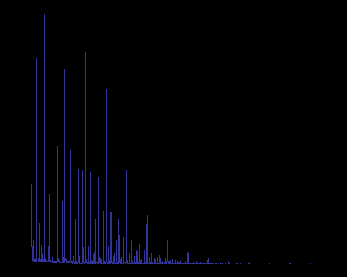
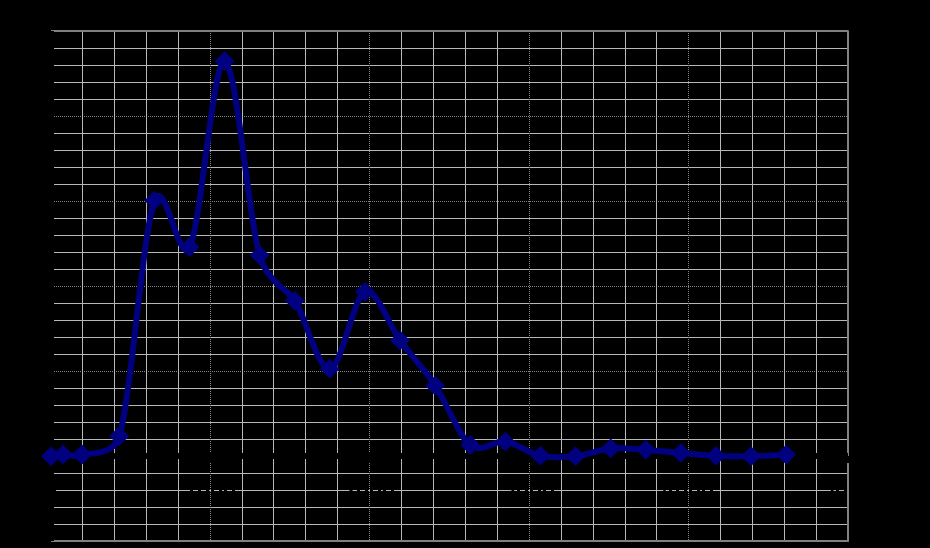
Масс-спектр и молекулярно-массовое распределение смеси олигомеров полиметиленнафталинсульфокислоты
Изучены спектры, полученные при применении α-циано-4-гидроксикоричной кислоты, 2,5-дигидроксибензойная кислота, синапиновой кислоты в качестве матриц и соли цезия в качестве катионизирующего агента.
Установлено предпочтительное применение α-циано-4-гидроксикоричной кислоты в качестве матрицы в связи с увеличением разрешающей способности метода относительно тяжелых фракций олигомеров.
По масс-спектрам рассчитаны значения среднечисловой и средневесовой молекулярных масс олигомера и его полидисперсности: MN = 1366,6 и ПД = 1,19.
Продолжается изучение влияния матрицы, типа катионизирующего агента и их соотношения на определение молекулярно-массового распределения олигомеров ПМНС.
ЭКСТРАКЦИОННО-СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ТЕОФИЛЛИНА В ВОДНЫХ СРЕДАХ
Кривошеева О.А.,1 Солохин С.А.2
1Воронежский государственный университет инженерных технологий,
Воронеж, Россия.
Аспирант 2г.
olesyakrivosheeva@yandex.ru
2Воронежский государственный университет инженерных технологий, Воронеж, Россия. Студент III курса.
Научный руководитель: Коренман Я.И.
Алкалоид теофиллин (1,3–диметилксантин), содержится в чае вместе с кофеином, применяется для лечения бронхиальной астмы. Известно, что теофиллин в больших дозах вызывает нервные расстройства и отравления. Для установления безопасности фармацевтических препаратов, содержащих теофиллин, необходима экспрессная и легковыполнимая методика, основанная, например, на жидкостной экстракции.
Известен коэффициент распределения теофиллина в системе хлороформ – вода (0,40), степень извлечения теофиллина при этом не превышает 3%. Такая система неприменима для решения практических задач, в частности, связанных с извлечением и концентрированием теофиллина.
Для повышения количественных характеристик экстракции (коэффициенты распределения D, степень извлечения R, %) применены гидрофильные растворители - алифатические спирты (н.пропиловый, н.бутиловый, н.пентиловый, н.гексиловый, н.гептиловый, н.нониловый) и изомерного (изопропиловый, изобутиловый, трет.бутиловый) строения.
Обязательное условие экстракции такими растворителями – насыщение водного раствора электролитом, понижающим растворимость распределяемых веществ в воде и обеспечивающим расслаивание системы. Высаливающие действие электролитов по отношению к органическим соединениям объясняется уменьшением содержания несвязанной воды в водном растворе. Высаливатель влияет на диэлектрическую проницаемость среды, ионную силу раствора, способствует обмену молекул воды из ближайшего окружения ионов экстрагируемого вещества на молекулы органического растворителя. В присутствии высаливателя образуется самостоятельная органическая фаза, повышаются количественные характеристики экстракции – коэффициентов распределения (D) и степени извлечения (R, %). Эффективность действия высаливателя зависит как от природы электролита, так и от свойств распределяемого вещества
Оптимальные параметры экстракции достигаются в системах с сульфатом аммония. Эта соль оказывает наибольшее высаливающее действие на экстракцию теофиллина по сравнению с другими изученными солями (хлорид натрия, сульфат натрия) вследствие более высокой растворимости в воде. Растворимость высаливателей увеличивается в ряду (г/100см3 воды, 200С):
Na2SO4 – NaCl – (NH4)2SO4
34,3 – 36,0 – 75,6
С повышением растворимости соли возрастает конкурирующее влияние соответствующих ионов. Это приводит к уменьшению гидратации, следовательно, растворимости экстрагируемого вещества в воде и усилению высаливающего действия.
Равновесную водную фазу после экстракции анализировали методом УФ-спектрофотометрии (SHIMADZU UV MINI-1240, кварцевая кювета, l = 1 см, λmax = 272 нм).
Коэффициенты распределения и степень извлечения теофиллина вычисляли по известным уравнениям:

% ,
,

где с0 и св – концентрации теофиллина в органической и водной фазах, мкг/см3; f – соотношение объемов равновесных водной и органической фаз.
Установлено, что в гомологическом ряду спиртов с возрастанием числа С−атомов их экстрагирующая способность закономерно снижается, что связано с уменьшением относительной доли полярных ОН−групп. Имеется прямая зависимость коэффициентов распределения теофиллина (lgD) от числа С-атомов в составе спиртов-гомологов. Зависимость описывается уравнением y = − 0,2869x + 2,3087, которое позволяет прогнозировать коэффициент распределения теофиллина, например, в системе с этиловым спиртом (D = 35,2).
Разработанный нами способ извлечения и определения теофиллина характеризуется экспрессностью и воспроизводимостью получаемых данных. Относительно невысокие коэффициенты распределения повышаются при повторной экстракции в идентичных условиях, степень извлечения достигает 92 %.
Работа выполнена в рамках ФЦП «Научные и научно-педагогические кадры инновационной России» (г/к № П2264 от 13.11.2009).
УСОВЕРШЕНСТВОВАНИЕ СПОСОБА ИДЕНТИФИКАЦИИ АМИНОКИСЛОТ ПО ЦВЕТНЫМ РЕАКЦИЯМ С ПРИМЕНЕНИЕМ ЦИФРОВЫХ ТЕХНОЛОГИЙ
Кудухова И.Г.,1 Рудакова Л.В.2
1Воронежский государственный архитектурно-строительный университет,
Воронеж, Россия.
Аспирант 3г.
i.n.g.a.85@mail.ru
2Воронежская государственная медицинская академия, Воронеж, Россия. Молодой учёный.
Научный руководитель: Рудаков О.Б.
Предложен способ цветометрической идентификации аминокислот с использованием цифрового фотоаппарата в качестве устройства, регистрирующего аналитический сигал в виде электронного изображения. Для оценки результатов цветометрических измерений по 2 цветным реакциям (биуретовой и реакции с нингидрином) предложены обобщенные показатели, представляющие собой лепестковые диаграммы с 6 осями, на которых отложены величины интенсивности цветовых компонент в цветовой модели RGB. Оценку интенсивности цветных реакций и компьютерную обработку цифрового изображения осуществляли с помощью графического редактора Adobe Photoshop (версия CS3).
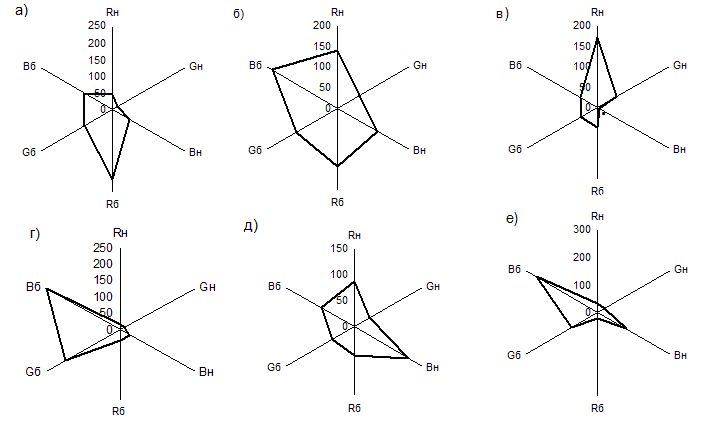
Рис. 1. Лепестковые диаграммы изменения цветности для различных аминокислот, С=5 г/л: а) L-аспарагин, б) L-лизин, в) глицин, г) глицил-глицин, д) DL-аланин, е) L-глутамин.
Как видно из рис.1, для каждой лепестковой диаграммы характерен индивидуальный профиль, следовательно, визуальная оценка профиля диаграммы может быть использована для качественной идентификации аминокислот. Для количественной оценки апробированы геометрические параметры диаграмм: площадь, периметр и фрактальность лепестковых диаграмм (рис.2, 3). Степень аппроксимации для градуировочных кривых (рис. 3) составляла R2≥0.98, что указывает на возможность их применения в количественном анализе.
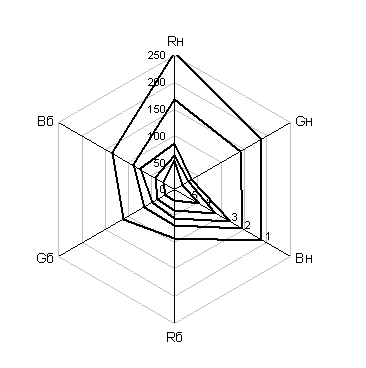
Рис. 2. Лепестковая диаграмма изменения цветности нингидриновой и биуретовой реакции в модели RGB для водных растворов DL-α-аланина различной концентрации: 1- 1,25; 2- 2,55; 3 - 5; 4 – 10; 5 – 20 г/л.
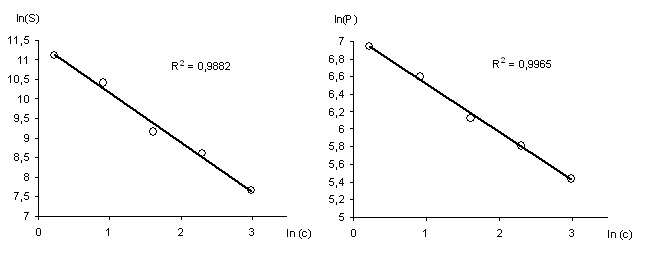
Рис.3. Калибровочные графики для определения DL-α-аланина по логарифмическим зависимостям площади (S) и периметра (P) диаграмм: ln(S)=11,4053(±0,0926)–1,2603(±0,0492)ln(c);
ln(Р)=7,0633(±0,0221)-0,5493(±0,0118)ln(c)
Таким образом, показана принципиальная возможность использования цветометрического метода и обобщенного идентификационного критерия в виде лепестковых диаграмм в качественном и количественном определении аминокислот в водных растворах.