
Синтез соединений на основе химических превращений производных α- оксокарбоновых кислот и их биологическая активность 15. 00. 02 фармацевтическая химия, фармакогнозия
| Вид материала | Автореферат диссертации |
- Синтез, свойства, биологическая активность n-гетериламидов α-оксокислот и продуктов, 367.21kb.
- Синтез, свойства и биологическая активность енаминоамидов ацилпировиноградных кислот, 439.81kb.
- Синтез и свойства новых производных 2 (1,2,4 триазолил 5 тио)уксусных кислот 15. 00., 240.9kb.
- Синтез, химические свойства и биологическая активность 1,4-дизамещенных 5-арил-3-гидрокси-3-пирролин-2-онов, 667.95kb.
- Синтез и биологическая активность карбо- и гетероциклов на основе тетрацианоэтилена, 673.64kb.
- Синтез, свойства и биологическая активность продуктов взаимодействия 1,2,4-триазолов, 669.98kb.
- Синтез, свойства и биологическая активность производных 2-хлорникотинонитрилов, 264.36kb.
- Реферат Синтез и превращения азотпроизводных карбоновых кислот, 346.86kb.
- Синтез и строение N,o- содержащих гетероциклических соединений на основе несимметричных, 201.05kb.
- Синтез, строение и реакции тиенилсодержащих кросс-сопряженных диеноновых производных, 287.71kb.
2. Синтез и строение N2-замещенных гидразидов α-оксокарбоновых кислот
2.1. Поиск методов синтеза N2-замещенных гидразидов 4-арил-2-гидрокси-4-оксо-2-бутеновых кислот
Нами установлено, что при взаимодействии 5-АФД 1 с этиловым эфиром гидразинэтановой кислоты образуются N2-этоксикарбонилметилгидразиды 4-арил-2-гидрокси-4-оксо-2-бутеновых кислот (8) и 3-арил-3,5-дигидрокси-2-этоксикарбонилметил-6-оксо-1,2,3,6-тетрагидропиридазины (9). Реакция протекает при эквимолярном соотношении реагентов при температуре 20-25 ˚С в среде диоксана:

Строение конечных продуктов реакции определяется характером заместителя в арильном фрагменте 5-АФД. Так, при введении электроноакцепторного заместителя равновесие двух форм сдвигается в сторону большего содержания линейных гидразидов 8а,б, в то время как, их кольчатая форма – пиридазиноны 9в,г выделяются из реакционной смеси с невысоким выходом. При введении электронодонорного заместителя гидразидов цепного строения не получено, а в качестве единственных термодинамически устойчивых соединений выделены пиридазиноны 9а,б.
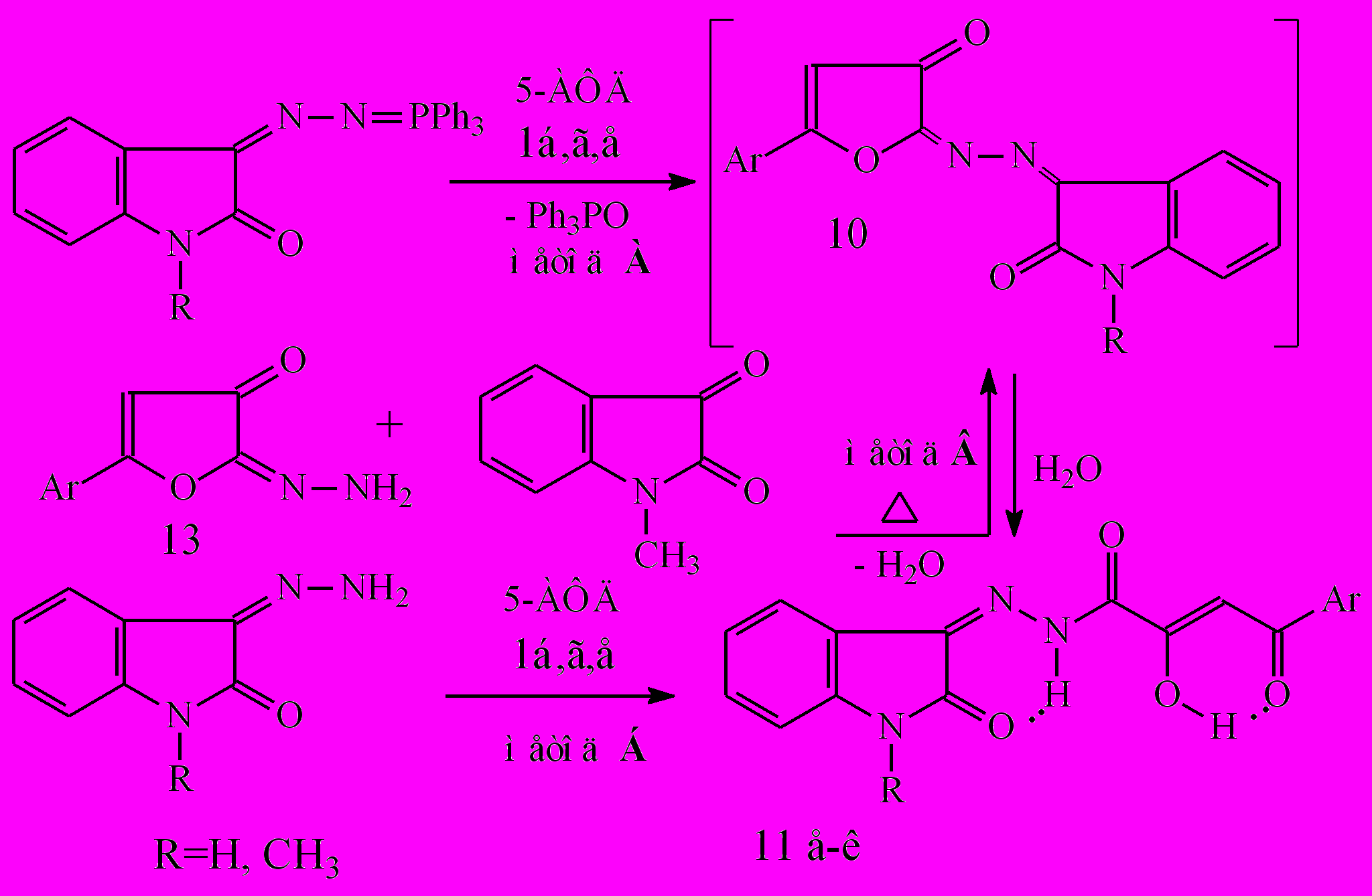
Ранее двухстадийным методом синтеза был получен небольшой ряд N2-замещенных метиленгидразидов АрПК и изучен их кислотный гидролиз. Однако биологическая активность исходных соединений и продуктов их гидролиза ранее не исследована. Представляло интерес расширить набор вводимых в реакцию с 5-АФД 1 фосфазинов для введения в структуру гидразидов биологически активных синтонов, изучить биологическую активность известных и не описанных ранее соединений, а также исследовать оптимальные условия проведения препаративного метода синтеза. Для этого первоначально реакцией Штаудингера при взаимодействии 5-АФД 1 с фосфазинами, полученными на основе бензоилдиазометана, дифенилдиазоацетона, адамантаноилдиазометана и этоксикарбонилдиазометана, синтезированы замещенные 2-метиленгидразоно-3-фураноны (10а-и). Их дальнейший кислотный гидролиз проводили при температуре 20-25°С в среде водного ацетонитрила при добавлении каталитических количеств 10% кислоты хлороводородной. В случае 3-фуранонов 10а-д реакция завершается образованием соответствующих N2-замещенных метиленгидразидов 11а-д (метод А). Соединения 11 г,д получены впервые. Замена диоксана на ацетонитрил при изучаемом гидролизе привела к значительному увеличению выхода целевых гидразидов.

10: R1=H, R2=C6H5CO, (C6H5)2СНСО, C10H15CO, C2H5CO
11: R1=H, R2=C6H5CO, (C6H5)2СНСО, С10Н15СО
Однако гидролиз 5-арил-2-этоксикарбонилметиленгидразоно-2,3-дигидро-3-фуранонов (10е-и) в аналогичных условиях останавливается на стадии присоединения воды с образованием 3-фуранонов 12а-г. Попытки выделить целевые метиленгидразиды 11 путем изменения условий эксперимента не увенчались успехом, что связано, по-видимому, с сильным электроноакцепторным влиянием этоксикарбонильной группы. В результате получены лишь известные 5-арил-2-гидразоно-2,3-дигидро-3-фураноны (13) и 3-арил-5-гидрокси-1,6-дигидро-6-пиридазиноны (14).
Установлено, что при взаимодействии 5-АФД 1 с фосфазинами, полученными на основе 1-R-3-диазо-2-оксоиндолов, направление реакции не меняется, в результате чего образуются соответствующие фураноны 10 и трифенилфосфиноксид (ТФО). Однако, по-видимому, вследствие их лабильности, в процессе выделения и очистки на первой стадии удается получить уже гидролизованные продукты – 1-R-2-оксо-3-индолинилиденгидразиды 11е-к. Вследствие этого, для оптимизации процесса гидролиза и сокращения времени при получение гидразидов 11е-к по методу А, нами предложен вариант без выделения промежуточных фуранонов 10. Гидразиды 11е-к получены также реакцией 5-АФД 1 с 3-гидразоно-1-R-2-оксоиндолами (метод Б). Двухстадийный метод синтеза В гидразидов 11 служит дополнительным химическим доказательством структуры гидразонов 13 и позволяет выделять азины 10, которые, зачастую, являются лишь интермедиатами в методе А, однако он является более трудоемким.
Важно отметить, что при получении некоторых метиленгидразидов 11 использование соответствующих фосфазинов является единственно возможным препаративным методом синтеза, в виду отсутствия или нестабильности исходных гидразонов.
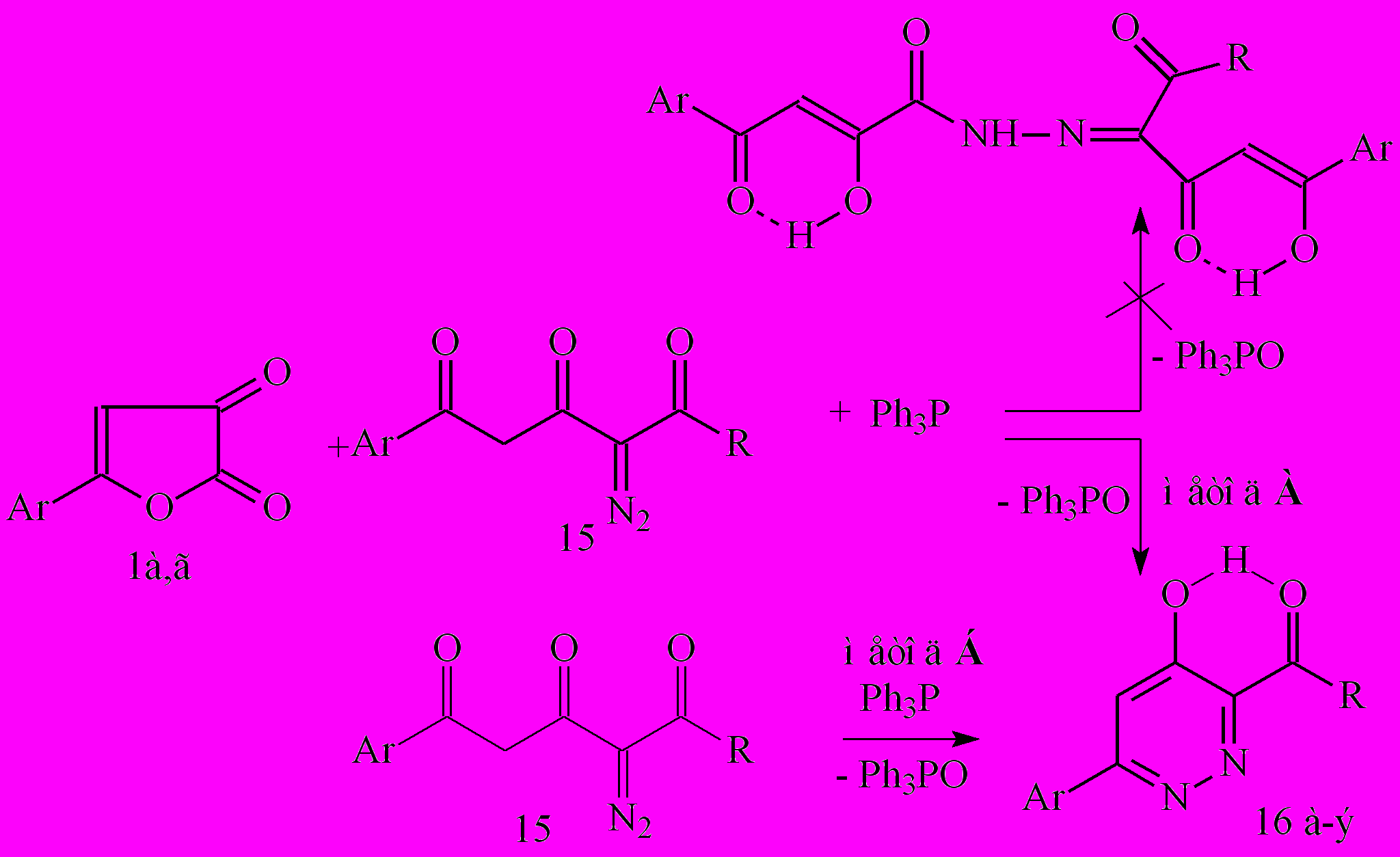
В литературе описан пример синтеза замещенного метиленгидразида АрПК на основе реакции 5-АФД с поликарбонильным диазосоединением в присутствии трифенилфосфина, позволяющий сочетать в молекуле два биологически активных ароилпирувоильных фрагмента. С целью синтеза таких гидразидов первоначально необходимо было получить исходные вещества реакцией ароилацетилирования диазокарбонильных соединений ароилкетенами (И2), полученными в результате термического декарбонилирования 5-АФД 1. Некоторые ароилдиазометаны и фталимидо-α-диазокетоны введены в реакцию впервые, в результате чего выделены новые 1,5-дизамещенные 2-диазо-1,3,5-пентантрионы (15и-о,р-у,ч-я):

15: R=OC2H5, C6H5, 4-NO2C6H4, 4-BrC6H4, 4-СН3C6H4, 2-СlC6H4, С10Н15, PhtCH2, PhtCH(СН3), PhtCH(С2Н5), Pht(CH2)2, Pht(CH2)3
Согласно данным спектров ЯМР1Н, в растворах ДМСО-d6, ДМСО-d6/CCl4 (1:3) соединений 15 присутствует как енольная форма Д (характеризуется уширенным сигналом протона енольного гидроксила при 15.32-15.62 м.д. и синглетом винильного протона при 6.91-7.19 м.д.), так и β-дикетонная форма Е (имеет синглет протонов метиленовой группы при 4.43-4.62 м.д.). Содержание последней составляет 21%-47% и зависит от характера заместителя R. Наличие двух форм в растворах диазопентантрионов 15 обуславливает особенности их химического поведения и определяет их дальнейшие химические превращения. Выходы соединений 15 и состав продуктов реакции зависят от характера заместителей как в ароматическом кольце ароилкетена, так и при диазокарбонильном фрагменте α-диазокетона, и колеблются от 16%-74%. Выявлено, что введение электронодонорных заместителей уменьшает содержание побочного продукта димеризации ароилкетена. Кроме того, при недостаточной нуклеофильности диазосоединений образующиеся производные 15 в условиях реакции ограниченно стабильны и могут претерпевать различные превращения.

16: R=OC2H5, C6H5, 4-NO2C6H4, 4-BrC6H4, 4-NO2C6H4, 4-CH3C6H4, 2-ClC6H4, C10H15, PhtCH(CH3), Pht(CH2)2, Pht(CH2)3
Нами установлено, что при взаимодействии 5-АФД 1с трифенилфосфином и диазопентантрионами 15, вопреки ожидаемым гидразидам АрПК, из реакционной смеси выделяются 3-замещенные 6-арил-4-гидроксипиридазины (16) и ТФО (метод А). Очевидно, процесс внутримолекулярной циклизации промежуточно образующегося фосфазина протекает быстрее, чем его взаимодействие с 5-АФД, что препятствует выделению фуранонов или гидразидов из реакционной смеси. Для наработки на биологические испытания пиридазины 16 были также получены прямым взаимодействием соединений по методу Б.
2.2. Синтез N2-замещенных метиленгидразидов 2-(2-гидроксифенил)-2-оксоэтановой кислоты
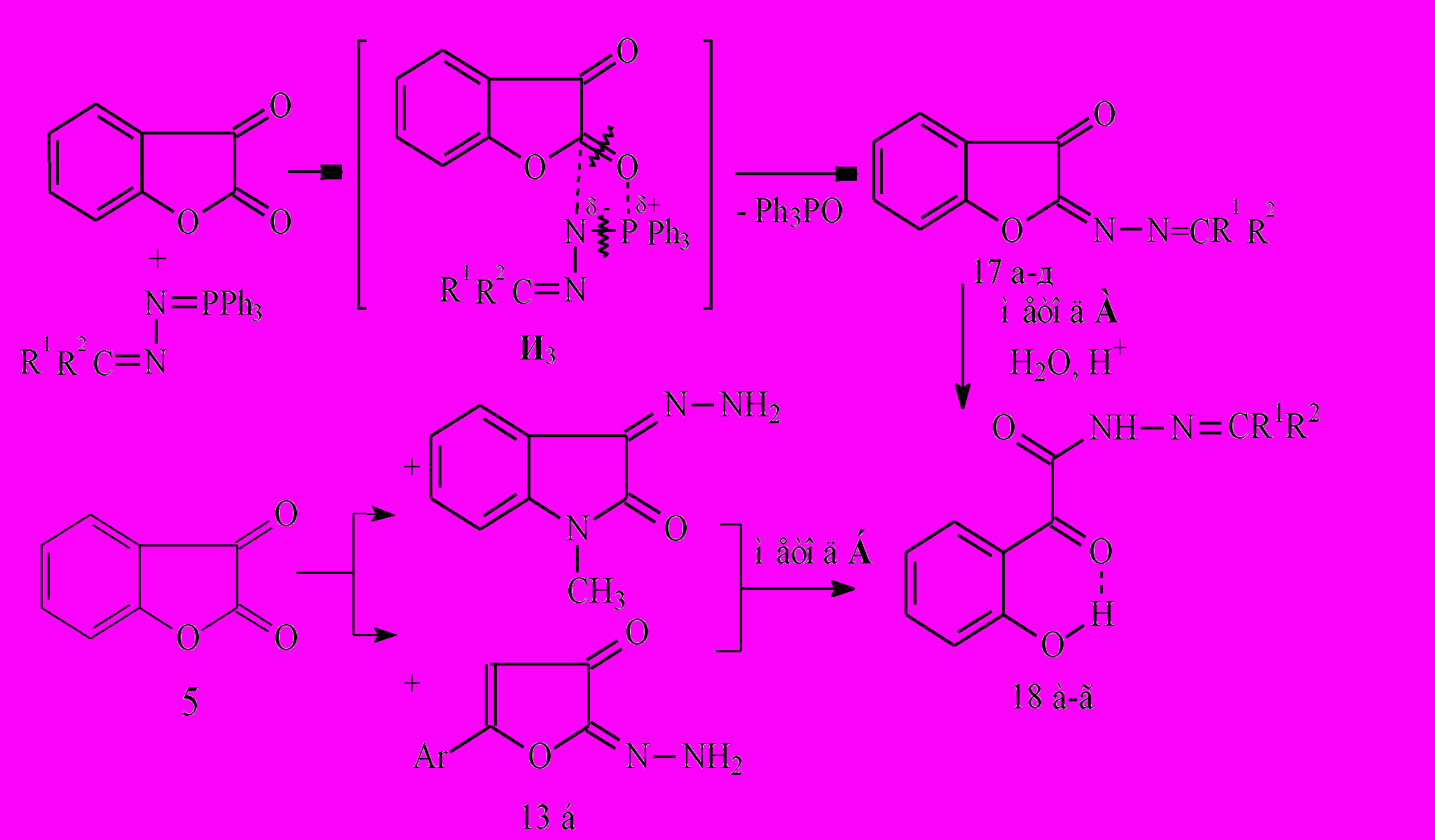
Нами впервые разработан двухстадийный метод синтеза N2-замещенных метиленгидразидов 2-(2-гидроксифенил)-2-оксоэтановой кислоты (18) через реакцию Штаудингера и последующий кислотный гидролиз образующихся 3-бензо[b]фуранонов 17 (метод А), позволяющий получать ранее недоступные соединения. Условия проведения реакций аналогичны получению производных АрПК 11. При наличии соответствующих гидразонов синтез гидразидов 18 может быть также одностадийным (метод Б). Также, как и гетериламиды 6, гидразиды 18, согласно спектральным характеристикам, существуют в кристаллическом состоянии и в растворе в форме с ВВС Н-хелатного типа.
 17: R1=R2=C6H5; СR1R2=C13H8 (9-флуоренилиден); R1=Н, R2=C6H5СО, C10H15СО; СR1R2=C9H7NO(1-метил-2-оксо-3-индонилиден). 18: R1=H, R2=C6H5CO, C10H15CO; CR1R2=C9H7NO
17: R1=R2=C6H5; СR1R2=C13H8 (9-флуоренилиден); R1=Н, R2=C6H5СО, C10H15СО; СR1R2=C9H7NO(1-метил-2-оксо-3-индонилиден). 18: R1=H, R2=C6H5CO, C10H15CO; CR1R2=C9H7NOМеханизм образования соединений 17, вероятно, аналогичен механизму взаимодействия фосфоранов с карбонильными соединениями и протекает, как и реакция Виттига, через четырехцентровое переходное состояние (И3). При этом полярность связи Р=N фосфазина в данной реакции играет определяющее значение.
3. Изучение химических свойств производных α-оксокарбоновых кислот и их γ-лактонных форм
3.1. Синтезы на основе производных 4-арил-2-гидрокси-4-оксо-2-бутеновых кислот
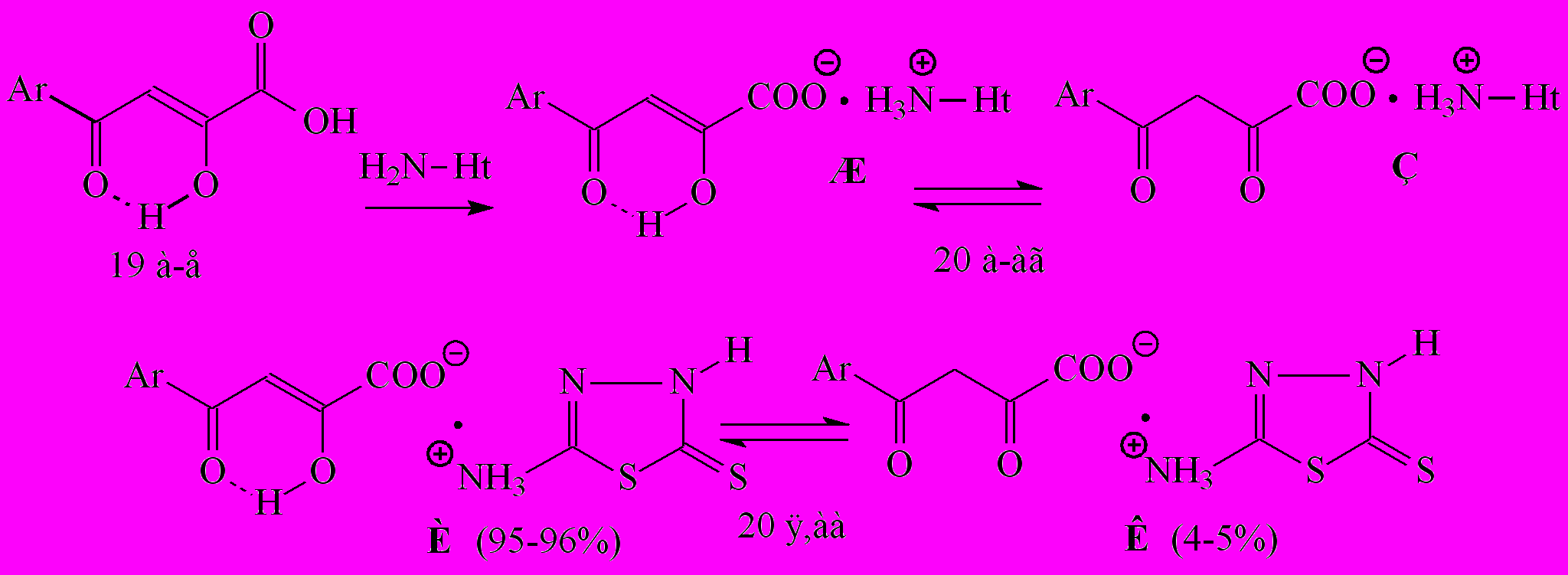
Ранее было показано, что при синтезе некоторых гетериламидов АрПК 2 на основе 5-АФД, в зависимости от строения гетероцикла и присутствия воды в реакционной смеси, в качестве сопутствующих соединений образуются соли АрПК. С целью получения биологически активных водорастворимых соединений нами изучено прямое взаимодействие АрПК 19 с гетериламинами различного строения, в результате чего выделены 4-арил-2-гидрокси-4-оксо-2-бутеноаты гетериламмония (20а-аг). Реакция протекает в безводном хлороформе при эквимолярном соотношении реагентов при температуре 20-25°С.
19: Ar=Ph(а), 4-СН3С6Н4(б), 4-СН3ОС6Н4(в), 4-С2Н5ОС6Н4(г), 4-СlС6Н4(д), 4-BrС6Н4(е). 20: Ht=3-C5H4N, 4-C5H4N, C5H3BrN, C3H2NS, C5H5N2 (4-метил-2-пиримидил), C2H1N2S, C3H3N2S, C4H5N2S, С2Н1N2S2, C7H5N2.
Как и у гетериламидов 2, во всех спектрах ЯМР1Н солей 20 присутствует синглет протонов метиленовой группы слабой интенсивности при 4.32-4.54 м.д., что свидетельствует о их существовании в растворе ДМСО-d6 в виде таутомерных форм Ж и З с преобладанием кетоенольной формы Ж (92-96%). Кроме того, подобно N-[2-(5-тиоксо-4,5-дигидро-1,3,4-тиадиазолил)]амиду АрПК 2аг, у бутеноатов 20я,аа в данном случае реализуется кето-енольная прототропная таутомерия тионной формы соединений, образующейся при миграции протона от сульфгидрильной группы к атому азота в положении 4 гетероцикла.
В продолжение изучения реакций АрПК с нуклефилами установлено, что этиловый эфир диазоэтановой кислоты взаимодействует с кислотами 19а-е региоселективно с образованием этоксикарбонилметиловых эфиров 4-арил-2-гидрокси-4-оксо-2-бутеновых кислот (21а-е). Реакция медленно протекает в среде спирта этилового при эквимолярном соотношении реагентов, при температуре 20-25°С и может быть полностью завершена при кипячении.

Согласно полученным спектральным характеристикам, соединения 21 енолизованы полностью, как АрПК 19 и их метиловые эфиры 3.
Нами исследовано взаимодействие N-[5-(6)-R-2-пиридил]амидов 2 с диазонуклеофилами различного строения. Установлено влияние заместителей гетерильной части этих амидов и степени нуклеофильности диазосоединения на состав и строение конечных продуктов реакции. Так, при введении в 5 положение атома брома исходных амидов в качестве основных продуктов реакции с диазометаном и диазоэтаном при соотношении реагентов 1:4 образуются α-О-алкильные производные 24б-ж, а арилдиазометаны и 9-диазофлуорен с ними не взаимодействуют. Введение в 6 положение гетероцикла метильной группы приводит к образованию сложной смеси соединений в реакции с диазометаном, среди которых выделены продукты гетероциклизации (соответствующие 2-оксоимидазо[1.2-а]пиридины 22а,б) и α-О-алкилирования (соединение 24а).

22: R1=СН3, R2=R3=H, Ar=Ph (а), 4-CIC6H4 (б); R1=H, R2=R3=Ph, Ar=Ph (в), 4-СН3C6H4 (г), 4-BrC6H4 (д), R1=СН3, Ar=4-СН3C6H4 (е); R1=H, R2=Ph, R3=4-BrC6H4, Ar=Ph (ж), 4-СН3C6H4 (з), R1=СН3, Ar=4-СН3C6H4 (и); R1=H, R2+R3=C12H8, Ar=Ph (л), 4-СН3OC6H4 (м). 24: R1=6-СН3, R2=R3=H, Ar=2,4-(СН3)2C6H3 (а); R1=5-Br, R2=R3=H, Ar=Ph (б), 4-СН3OC6H4 (в), 4-ClC6H4 (г), 4-BrC6H4 (д); R1=5-Br, R2=H, R3=СН3, Ar=Ph (е), 4-СН3OC6H4 (ж). 25: Ar=Ph (а), 4-С2Н5OC6H4 (б)
Кроме того, из реакционной смеси гетериламида 2е с диазометаном, наряду с соединением 22а, был получен N-(6-метил-2-пиридил)амид 3-бензоил-4-гидрокси-4,5-дигидропиразол-4-карбоновой кислоты (23). Образование пиразола 23 происходит в результате реакции 1,3-диполярного циклоприсоединения диазометана по кратной связи С2=С3 исходного пиридиламида.
При взаимодействии N-(6-метил-2-пиридил)амидов 2 с диазоэтаном основным процессом является 1,3-диполярное циклоприсоединение диазонуклеофила, в результате чего выделены соединения 25а,б, а в случае с диарилдиазометанами – гетероциклизация в соответствующие производные имидазо[1.2-а]пиридинов 22в-и. В реакции амида 2е с дифенилдиазометаном было получено также производное 22к, у которого, в отличие от соединений 22а-и, карбонильная группа бензоильного заместителя енолизована нацело, о чем свидетельствуют его характеристики.
При использовании диазофлуорена в реакции с N-(2-пиридил)амидами 2а,в основным процессом является гетероциклизация в соединения 22л,м, однако для этого требуются более жесткие условия эксперимента.
Образование имидазо[1,2-а]пиридинов 22 начинается, по-видимому, с протонирования диазонуклеофила атомом водорода енольного гидроксила и перегруппировки 2-пиридиламидного фрагмента в пиридоимидный с образованием интермедиата И4. Создание имидазольного цикла сопровождается внедрением диазокарбокатиона по кратной связи С2=С3, элиминированием азота и миграцией атома водорода к атому С3:
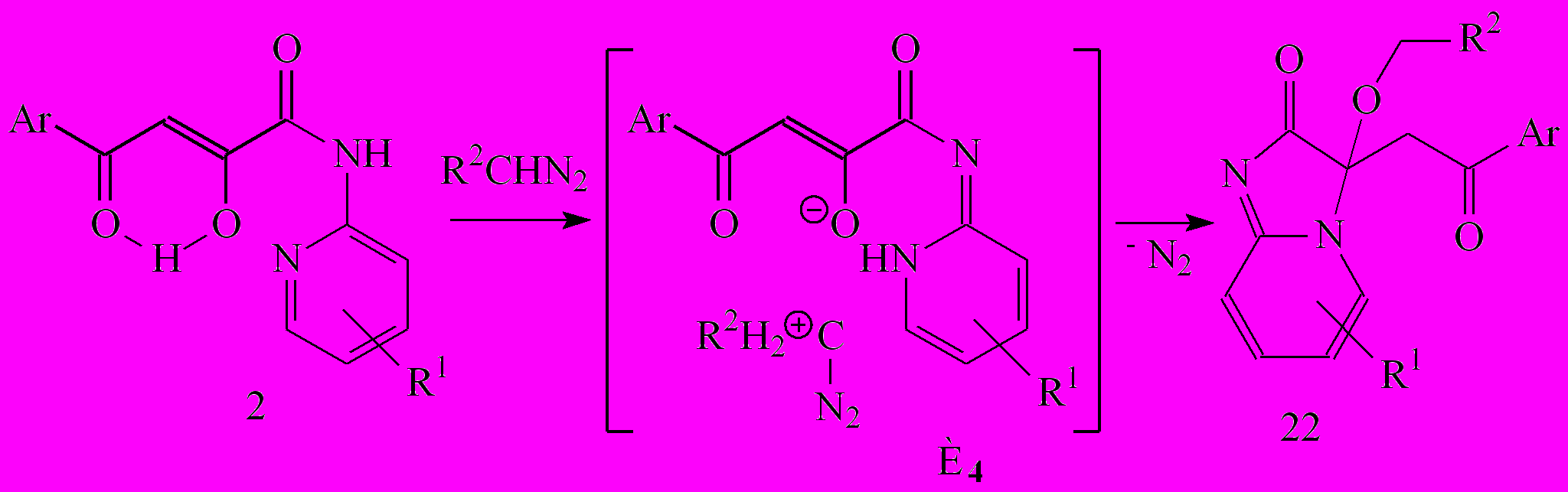
Попытка синтеза водорастворимых гидрохлоридов на основе имидазопиридинов 22в,к не удалась, поскольку исходные соединения дециклизуются с образованием соответствующих гидрохлоридов N-[6-R-(2-пиридил)]амидов 4-арил-2-гидрокси-4-оксо-2-бутеновых кислот (26а,б).
Установлено, что N-(5-R-2-тиазолил)амиды 2 внутримолекулярно циклизуются в производные имидазо[2,1-b]тиазолов 27,28 только под действием диазометана и диазоэтана. При взаимодействии N-(2-тиазолил)амида 2у с диазоэтаном был также выделен N-(2-тиазолил)амид 4-оксо-4-фенил-2-этокси-2-бутеновой кислоты (29). При проведении реакции N-(2-тиазолил)амида 2ф с диазоэтаном в аналогичных условиях получен продукт 1,3-диполярного циклоприсоединения – N-(2-тиазолил)амид 3-(4-хлорбензоил)-5-метилпиразол-4-карбоновой кислоты (30).
Взаимодействие эквимолярных количеств гетериламида 2у с дифенилдиазометаном при кипячении реакционной смеси приводит к образованию продукта β-С-алкилирования исходного амида – соединению 31, что вероятно, можно объяснить [1,3]-сигматропной перегруппировкой первоначально образующихся продуктов С2-О-дифенилметилирования исходного амида в реакционных условиях (110оС), вследствие большей термодинамической устойчивости соединения 31:
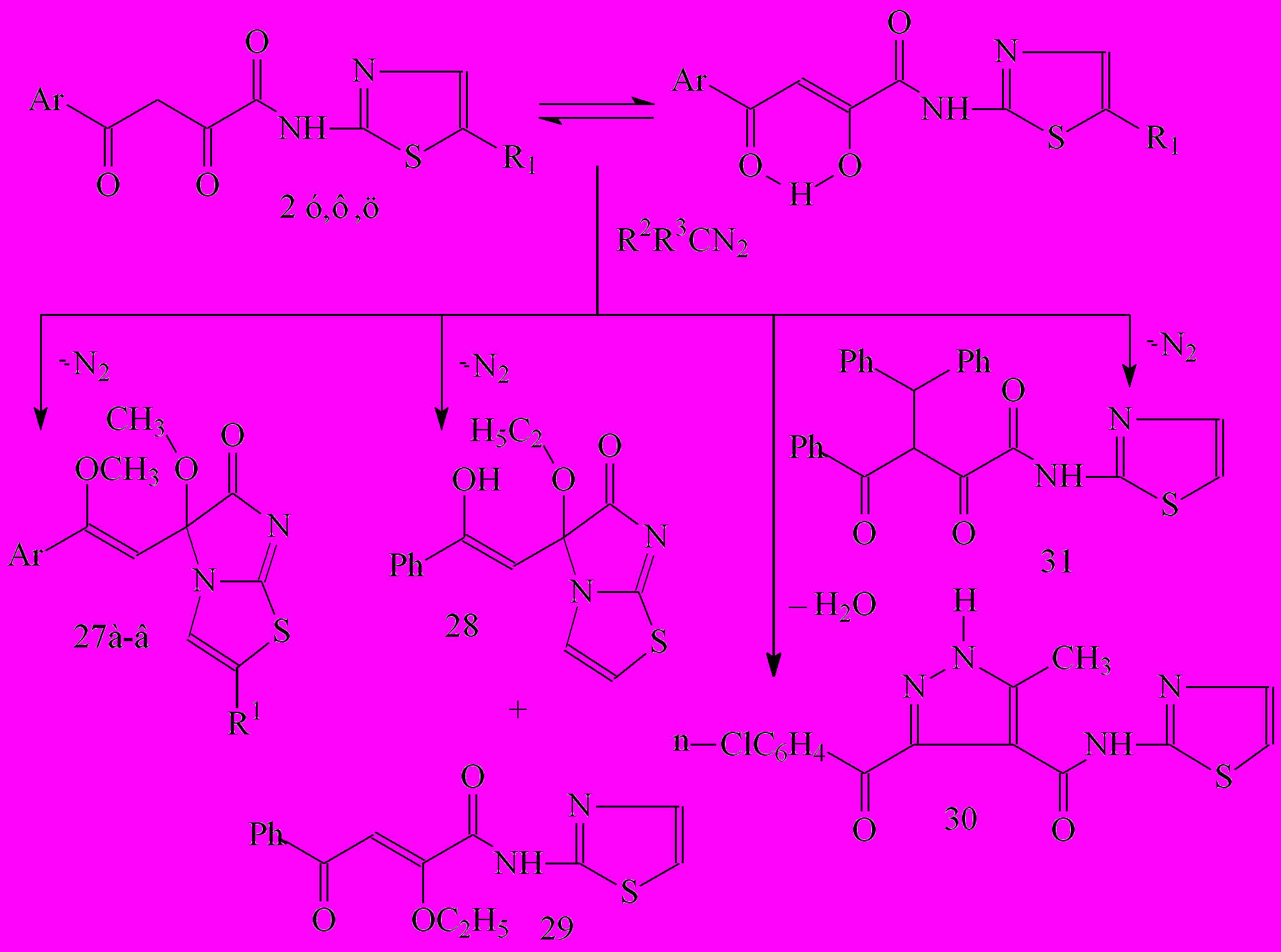
27: R1=H, Ar=C6H5 (а), 4-ClC6H4 (б); R1=NO2, Ar=C6H5 (в)
Реакция бензоилметиленгидразидов АрПК 11 с диазометаном региоселективна и приводит к α-О-метильным производным 32а,б. Их спектральные характеристики хорошо согласуются с данными аналогичных производных N-гетериламидов 24, 29.

32: Ar=4-СH3C6H4 (а), Ar=4-СlC6H4 (б)
В продолжение исследования реакции Штаудингера, позволяющей вводить в карбонильный субстрат потенциально биологически активные азофрагменты, нами изучены реакции АрПК 19, метилового эфира АрПК 3 и анилидов АрПК 33 с трифенилфосфазинами различного строения. Установлено, что вне зависимости от структуры карбонильного соединения, образуются продукты по α-карбонильной группе его дикетоформы - соответственно замещенные 2-метиленгидразино-4-арил-4-оксо-2-бутеновые кислоты (34а-з), анилиды 34о-с и метиловый эфир 34т (метод А). Производные 34и-н получены методом Б – реакцией кислот 19 с моногидразоном бензила. Полученные спектральные характеристики соединений 34 свидетельствуют о существовании их в кристаллическом состоянии и в растворе ДМСО-d6 в енгидразинной форме с ВВС Н-хелатного типа с локализацией атома водорода при атоме азота, т.е. в форме Z-изомера.

3: Х=СН3О, R1=Br (в). 33: Х=NHC6H5, R1=H (а), CH3 (б), Cl (в)
34: Х=ОН, R2=R3=C6H5; R2=H, R3=C6H5СО; R2=C6H5, R3=C6H5СО; Х=NHC6H5, R2=R3=H; R2=Н, R3=C6H5СО; Х=CH3О, R2=R3= C6H5
При проведении реакции кислоты 19а, эфира 3а и анилида 33а с трифенилфосфазином, полученным на основе этоксикарбонилдиазометана, в аналогичных условиях из реакционной смеси удается выделить лишь ТФО и известные производные 3-фенил-5-пиразолкарбоновой кислоты (35а-в).
Нами установлено, что N-гетериламиды АрПК 2 взаимодействуют с гидразонами бензофенона, фенил(4-бромфенил)кетона и 9-флуоренона с образованием соответственно N-гетериламидов 4-арил-2-диарилметиленгидразино- и 4-арил-2-флуоренилиденгидразино-4-оксо-2-бутеновых кислот (36,37):
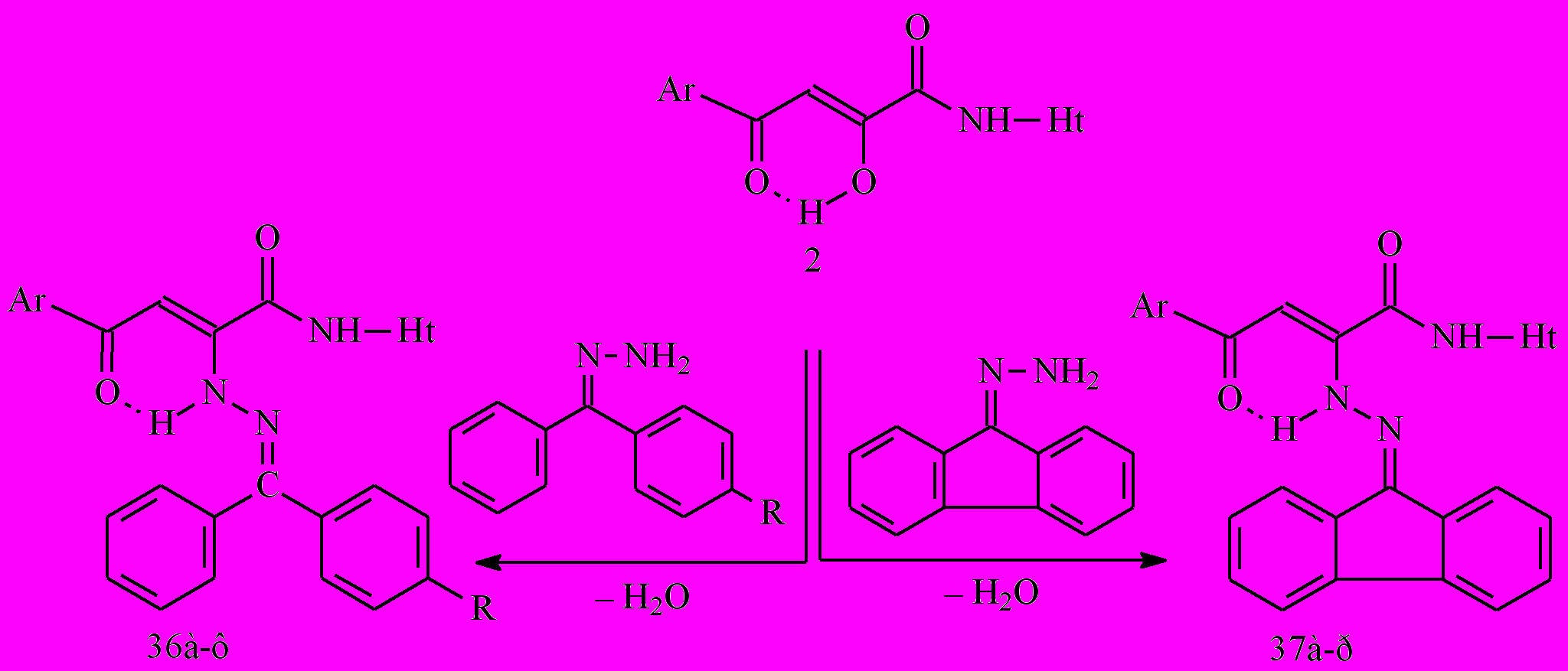
R=Н, Br. 36: Ht=2-С5Н4N, С6Н6N, 3-С5Н4N, С3Н2NS, С2Н1N2S, С3Н3N2S, С4Н5N2S, С11Н11N2O, С7Н5N2.
37: Ht=2-С5Н4N, 3-С5Н4N, С3Н2NS, С2Н1N2S, С3Н3N2S, С4Н5N2S, С11Н11N2O.
Изучение спектров ЯМР1Н соединений 36 и 37, снятых в ДМСО-d6 и CDCl3, показало наличие Z-кетоенгидразинной ( Z-KEГ), Е-кетоенгидразинной (Е-КЕГ) и -кетогидразонной (-КГ) форм в растворе этих соединений. Нами установлено, что состав таутомерных форм в растворе определяется, в первую очередь, природой растворителя, а также строением гетероциклического и гидразинного фрагментов. Так, в растворе ДМСО-d6 соединений 36,37 форма Z-КЕГ наблюдается у всех изученных веществ, составляет 32-100% для соединений 36 и 15-80% для соединений 37.
Содержание минорной формы Е-КЕГ в растворе ДМСО-d6 составляет 25-48% для производных 36 и 15-28% для производных 37. Она характеризуется сдвигом сигнала протона NH-группы, вовлеченного во ВВС, в более слабое поле до 8.45-11.22 м.д., а также небольшим сдвигом сигналов протонов NH-N= и –СН= групп в сильное поле, где они наблюдаются, соответственно, при 9.55-13.35 и 6.12-6.67 м.д..

Для формы β-КГ характерен синглет двух протонов метиленовой группы при 4.42-4.62 м.д. и отсутствие сигнала протона –NH-N= группы. При этом её содержание для производных 36 составляет 15-50%, а для производных 37 – 50-100%. В целом, форма Е-КЕГ присутствует в таутомерном равновесии трех форм только в растворе ДМСО-d6, а в растворе CDCl3 – только в равновесии с Z-КЕГ со значительным преобладанием последней. Введение электроноакцепторного заместителя в ароильный фрагмент соединений 36,37 приводит к сдвигу таутомерного равновесия в сторону большего содержания формы Z-КЕГ только в растворах ДМСО-d6.
Нами установлено, что при взаимодействии N-гетериламидов АрПК 2 с этиловым эфиром гидразинэтановой кислоты направление реакции не меняется, но осуществляется дальнейшее замыкание в цикл с образованием N-гетериламидов 5-арил-1-этоксикарбонилметил-1Н-пиразол-3-карбоновых кислот (38).
При изучении спектров ЯМР1Н производных N-[2-(5-R-1,3,4-тиадиазолил)]амидов 38б-з в ДМСО-d6 и СDCl3, нами обнаружена прототропная амино-иминная таутомерия - наличие соответствующих таутомерных форм И и К. Таутомерное равновесие этих форм зависит от природы растворителя, а также от характера заместителя в Ar и Ht. Так, при снятии спектров ЯМР1Н соединений 38г,д в разных растворителях установлено, что при введении заместителя в арильный фрагмент наблюдаются обе формы с преобладанием формы К в ДМСО-d6 и формы И в СDCl3, в то время как отсутствие заместителя способствует их 100% содержанию в соответствующем растворителе. При анализе спектров амидов 38в-з, снятых в ДМСО-d6, выявлено, что соединения 38г,е существуют в форме К (100%), в то время как их замещенные аналоги 38в,д,ж,з существуют в виде равновесных форм К:И (43-62% : 38-57%), количественное содержание которых определяется, в свою очередь, строением гетериламидного фрагмента.


38: Ht=3-С5Н4N, С2Н1N2S, С3Н3N2S, С4Н5N2S
Механизм образования соединений 38, по-видимому, включает первоначальную атаку первичной аминогруппы замещенного гидразина на карбонильную группу С2=О субстрата. Образующийся при этом интермедиат И5 элиминирует воду и циклизуется в производное пиразола И6 за счет внутримолекулярной атаки вторичной аминогруппы гидразина на карбонильную группу С4=О субстрата, а последующее отщепление молекулы воды приводит к соответствующим соединениям 38.
Реакция N-гетериламидов АрПК 2 с катионами переходных металлов ранее не исследована. Эти соединения могут быть использованы в качестве лигандов, благодаря наличию нескольких потенциально активных комплексообразующих групп β-дикарбонильного, амидного и гетероциклического фрагментов. Нами установлено, что амиды 2 взаимодействуют с дихлоридами меди, цинка, кадмия в соотношении 2:1, в мягких условиях в среде спирта этилового с образованием бис[3-арил-1-(N-гетерил)карбоксамидо-1,3-пропандионато] меди (39а-о), цинка (40а-л), кадмия (41а-л):

39: Me=Сu, Ht=2-С5Н4N, С5Н3BrN, 3-С5Н4N, С3Н2NS, С2Н1N2S, С3Н3N2S, С4Н5N2S, С11Н11N2О 40: Ме=Zn, Ht=2-С5Н4N, С5Н3BrN, 3-С5Н4N, С3Н2NS, С2Н1N2S, С3Н3N2S, С7Н5N. 41: Ме=Сd, Ht=2-С5Н4N, 3-С5Н4N, С3Н2NS, С3Н3N2S, С4Н5N2S, С7Н5N2
Полученные результаты атомно-абсорбционного анализа комплексных соединений свидетельствуют о том, что металл в составе комплекса связан с двумя лигандами, а отрицательная проба Бельштейна показывает также отсутствие атомов хлора. По-видимому, хлор элиминируется из зоны реакции в составе молекулы HСl.
На наш взгляд, для соединений 39-41 наиболее вероятными структурами продуктов реакции являются Л, М, Н. Кроме того, не исключаются структуры с возможными координационными связями с гетероатомами в цикле.
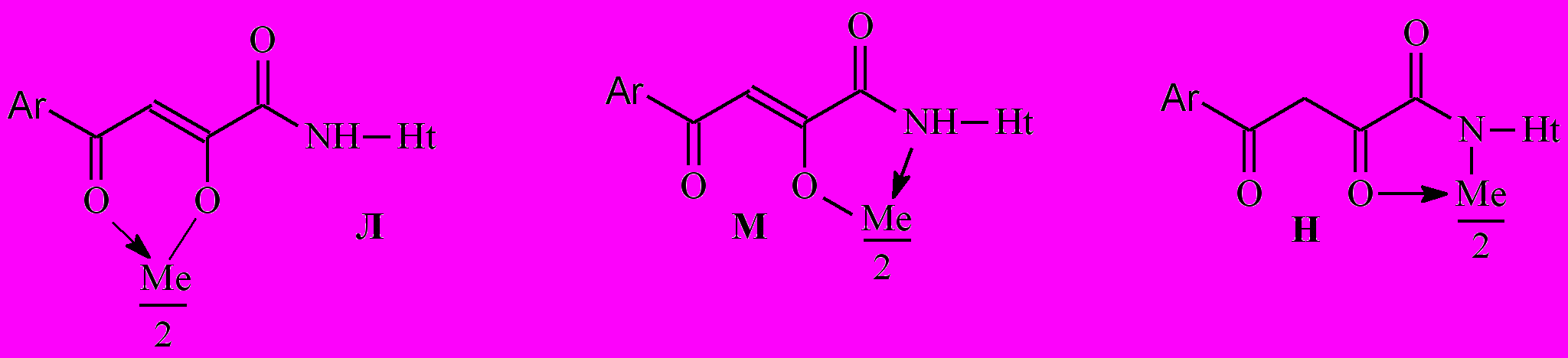
В пользу структуры Л свидетельствует наличие полос поглощения NH-групп в ИК-спектрах соединений 39-41, а также наличие сигнала протонов NH-групп в спектрах ЯМР1Н. Кроме того, в ИК-спектрах полоса поглощения С4=О смещена в область низших частот на 10-15 см-1, по сравнению с исходными амидами, что свидетельствует об её участии в образовании более прочной координационной связи.

Для подтверждения структуры хелатов нами выполнены квантово-химические расчеты неэмпирическим методом в базисе G6 31* в приближении АМ1 у соединения 40а для четырех комплексов цинка, ковалентно связанного с двумя лигандами через атомы кислорода О2 енолизованных карбонильных групп и имеющего соответствующие координационные связи (структуры О, П, Р, С). Согласно выполненным расчетам и вычисленным значениям теплот образования комплексов (ΔНf), наиболее стабильной является форма комплекса П. Таким образом, N-гетериламиды 2 в реакции комплексообразования координируются металлами как бидентатные О-О лиганды и образуют с ними продукты со структурой типа П с делокализованными кратными связями в шестичленных хелатных циклах.
Нами установлено, что направление реакции комплексообразования не меняется при использовании в качестве исходных соединений N2-замещенных метиленгидразидов 11а,в и дихлоридов меди, цинка, кадмия при соотношении реагентов 2:1. Взаимодействие протекает в аналогичных вышеописанным условиях с образованием бис {3-арил-1-[N2-(бензоилметилен)карбоксгидразидо]-1,3-пропандионато}меди, цинка, кадмия (42а-д):

42: Me=Cu, R=CH3 (а), Сl (б); Me=Zn, R=Cl (в); Me=Сd, R=CH3 (г), Сl (д)
Спектральные характеристики соединений 42 согласуются с данными, полученными для хелатов 39-41.