
Лекция 1 систематическая номенклатура (ганча-видмана) моноциклических соединений. Номенклатура конденсированных систем
| Вид материала | Лекция |
- Номенклатура органических соединений. Номенклатура – совокупность правил составления, 82.43kb.
- Номенклатура полимеров и сополимеров, 89.7kb.
- Систематическая номенклатура непредельные углеводороды, 30.7kb.
- 1. Изомерия и номенклатура, 41.98kb.
- Разграфка и номенклатура карт разграфка карт, 55.1kb.
- Методические рекомендации по применению номенклатуры дел Южного федерального университета, 60.7kb.
- Порядок составления номенклатуры дел, 135.26kb.
- Лабораторная работа №2 Важнейшие классы неорганических соединений, 88.03kb.
- План занятия №1 Классификация, номенклатура и пространственное строение органических, 96.42kb.
- 1. Номенклатура медицинских услуг (далее Номенклатура) представляет собой перечень, 6016.43kb.
Лекция 1
СИСТЕМАТИЧЕСКАЯ НОМЕНКЛАТУРА (ГАНЧА-ВИДМАНА) МОНОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ. НОМЕНКЛАТУРА КОНДЕНСИРОВАННЫХ СИСТЕМ.
ПРИНЦИП. Название гетероцикла строится путем объединения префикса, обозначающего присутствующие гетероатомы и корня, указывающего на размер цикла и его насыщенность.
Префиксы:
Элемент | Валентность | Префикс |
| Кислород | 2 | Окса |
| Сера | 2 | Тиа |
| Селен | 2 | Селена |
| Азот | 3 | Аза |
| Фосфор | 3 | Фосфа |
| Мышьяк | 3 | Арса |
| Кремний | 4 | Сила |
| Бор | 3 | Бора |
^
Корни слов
| Число атомов в цикле | Ненасыщенный цикл | ^ Насыщенный цикл |
| 3 | ирен (ирин*) | иран (иридин*) |
| 4 | ет | етан (етидин*) |
| 5 | ол | олан (олидин *) |
| 6 | ин | инан (ан**) |
| 7 | епин | епан |
| 8 | оцин | окан |
| 9 | онин | онан |
| 10 | ецин | екан |
| ||
Если в молекуле присутствуют два и более гетероатома, префиксы перечисляются в том порядке, в каком они приведены в таблице префиксов (см. выше). Число одинаковых атомов указывается числовыми префиксами: ди-, три-, тетра и т.д.
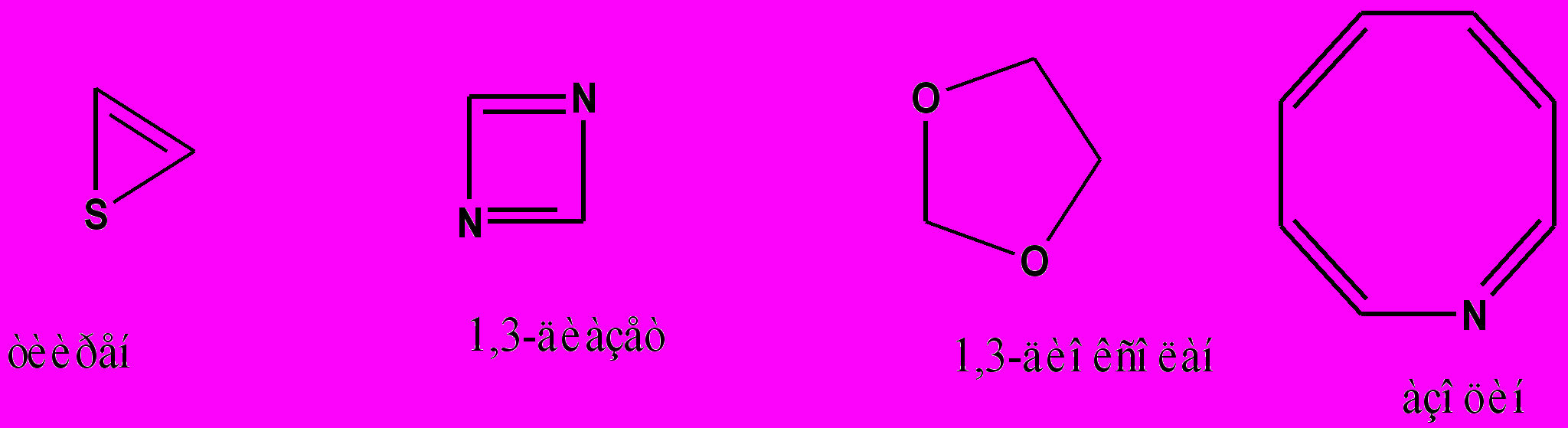
Если в циклической системе, при наличии максимального числа двойных связей все же есть насыщенный атом, положение этого атома обозначается цифрой с префиксом H – как часть названия гетероциклической системы. Этому (называемому “определяемым”) положению присваивают наименьший номер.
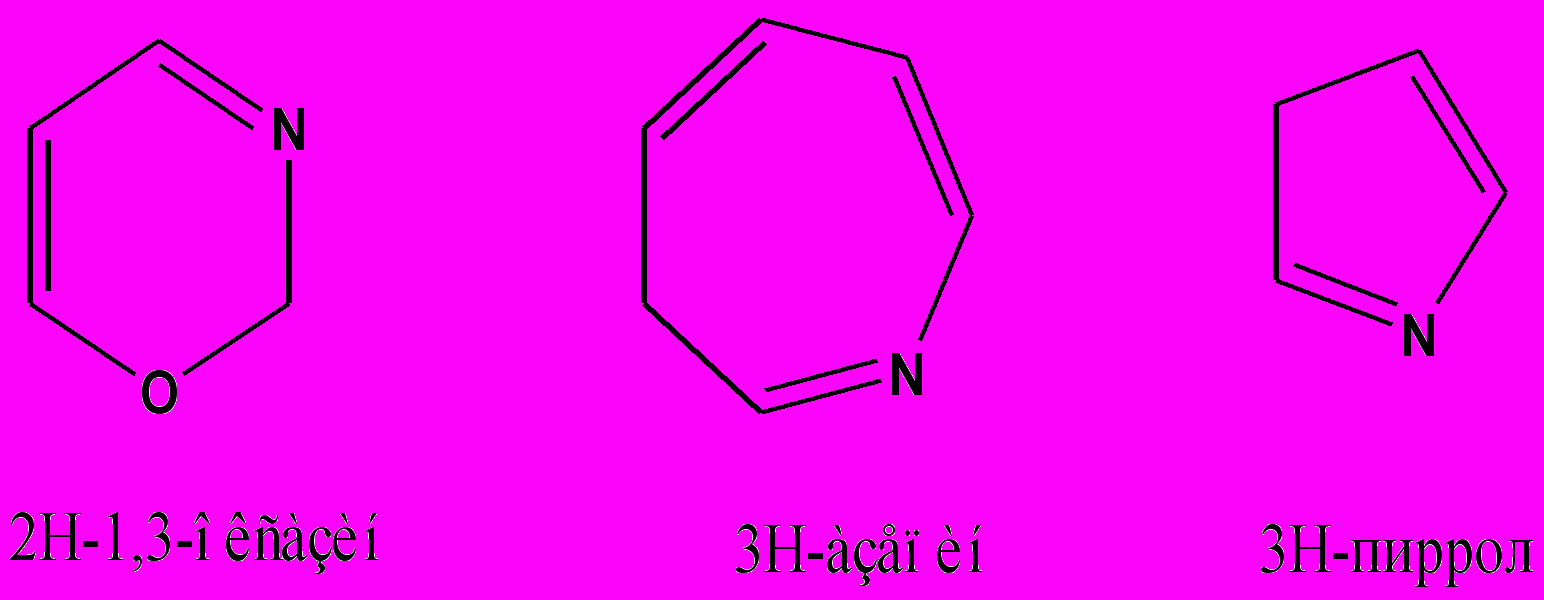
Эти названия не заменяют признанных тривиальных – например пиррол не называют азолом.
^
Номенклатура конденсированных систем
Подавляющее число гетероциклических соединений содержит два и более конденсированных цикла.
ПРИНЦИП. При наименовании конденсированных гетероциклических систем их “разбивают” на составные части.
- Названия компонентов гетероциклической системы выбирают из списка “признанных” тривиальных названий. Необходимо выбрать “больший” компонет (например хинолин, а не пиридин). Если моноциклический компонент не имеет тривиального названия, его называют по системе Ганча-Видмана.
- Необходимо выбрать один из компонентов в качестве “основного” (см. схему ниже)
- Второй компонент добавляется к основному в виде префикса (название цикла+ буква о) (пиример: пиррол – пирроло) .
Исключения:
| Изохинолин | Изохино |
| Имидазол | Имидазо |
| Пиридин | Пиридо |
| Тиофен | Тиено |
| Фуран | Фуро |
- Связи, образующие циклическую систему основного компонента обозначают буквами латинского алфавита, начиная со стороны, обычно имеющей номер 1,2. Атомы циклической системы второго компонента нумеруются обычным образом. Место сочленения циклов обозначается соответствующими буквой и цифрами, причем порядок цифр должен быть выбран таким образом, чтобы стрелки изображенные на рисунке шли навстречу друг другу.
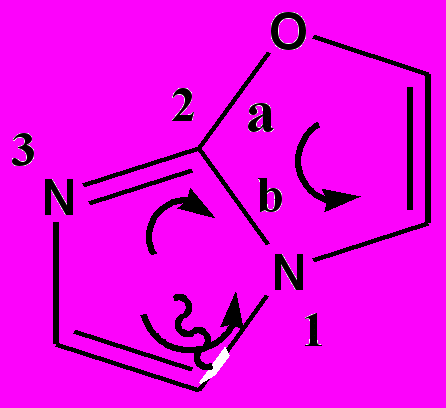 Основной компонент – оксазол (сторона “b”), второй компонент – имидазол – сторона 1-2 – сочленение - [2,1-b] – имидазо[2,1-b]оксазол
Основной компонент – оксазол (сторона “b”), второй компонент – имидазол – сторона 1-2 – сочленение - [2,1-b] – имидазо[2,1-b]оксазол- Нумерация в конденсированных системах проводится независимо от вышеперечисленных действий и обычно начинается с атома, ближайшего к сочленению циклов, но так, чтобы у гетероатома был наименьший номер. В альтернативных случаях наименьший номер должен иметь “старший” гетероатом и сумма индексов гетероатомов должна быть наименьшей.
Лекция 2
Трехчленные гетероциклы с одним гетероатомом. Азиридин, оксиран, тииран,
^
Методы синтеза
- Азиридин
Самым распространенным методом синтеза является присоединение нитренов и нитреновых ионов к алкенам и алкинам , достаточно часто также используют реакцию присоединения карбенов к иминам и нитрилам. Нитрены обычно синтезируют окислением различных N-аминогетероциклов тетраацетататом свинца (Схема 1). Описан также синтез на основе S-стабилизированного нитрена (Схема 2).
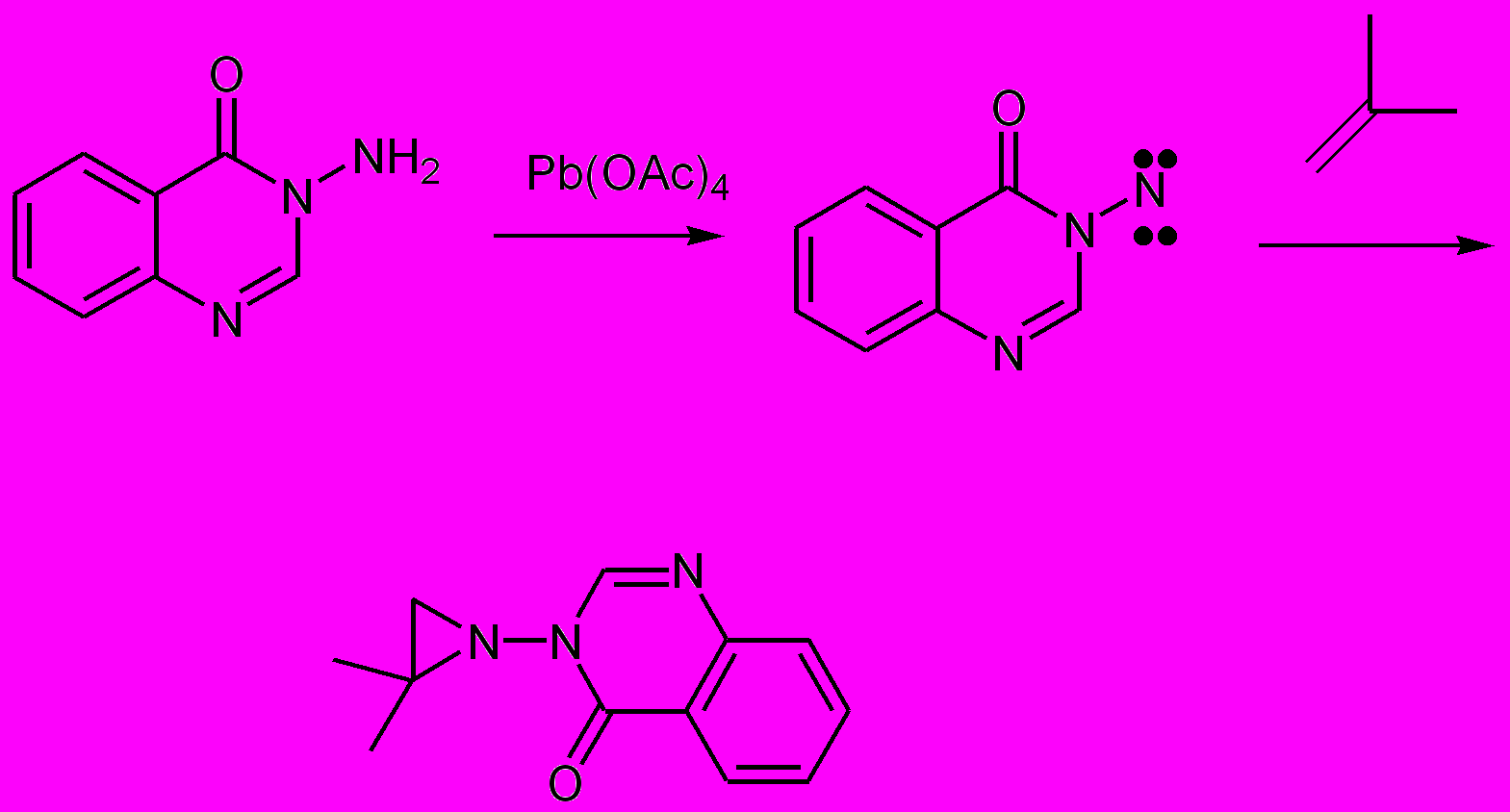
Схема 1
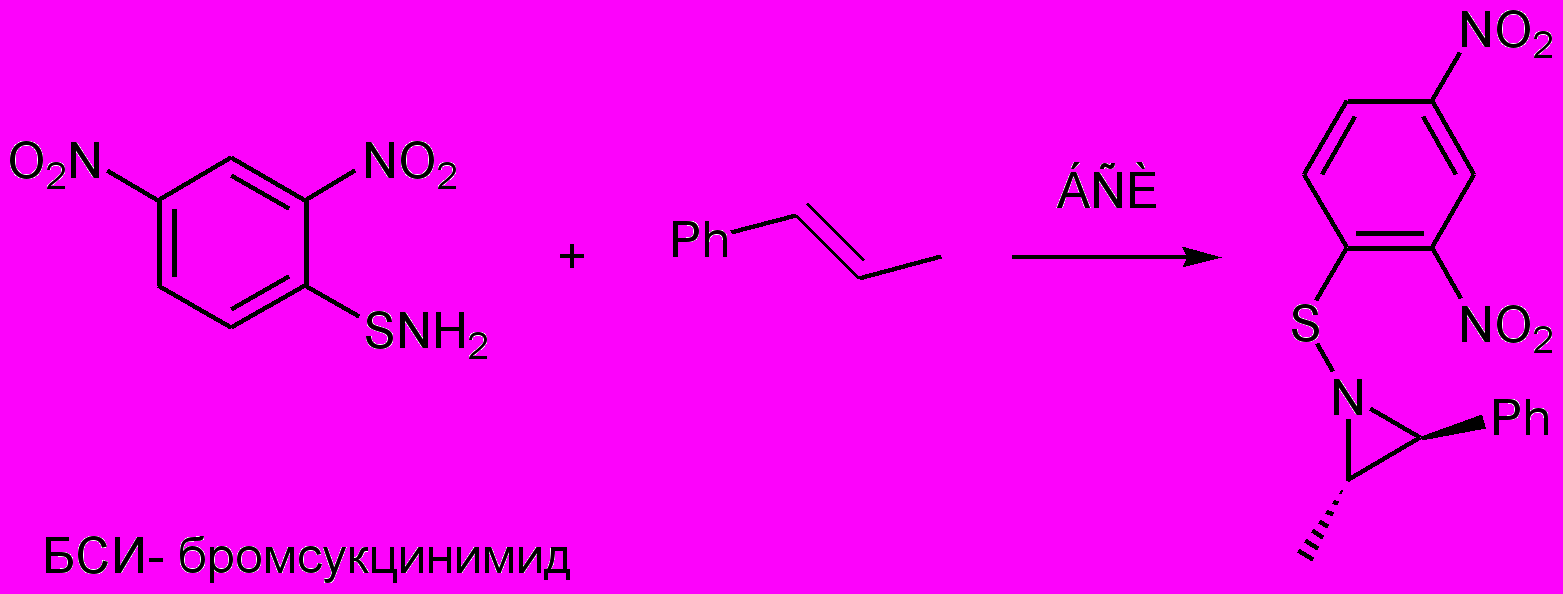
Схема 2
- Оксиран
Оксираны могут быть получены путем прямого окисления алкенов надкислотами (реакция Прилежаева) или кислородом в присутствии катализатора. Наиболее часто используют м-хлорнадбензойную кислоту. Реакция облегчается при наличии алкильных заместителей при дойной связи алкена и напротив затрудняется в случае наличия электроно-акцепторных заместителей. Обычно окисление надкислотами проходит стереоспецифично. Энантиоселективное эпоксидирование алилловых спиртов достигается использованием в качестве катализатора смеси соли Ti 4+и оптически активного диэтилтартрата (Схема 3) При замене одного энантиомера тартрата на другой происходит селективное образование другого энантиомера эпоксида.
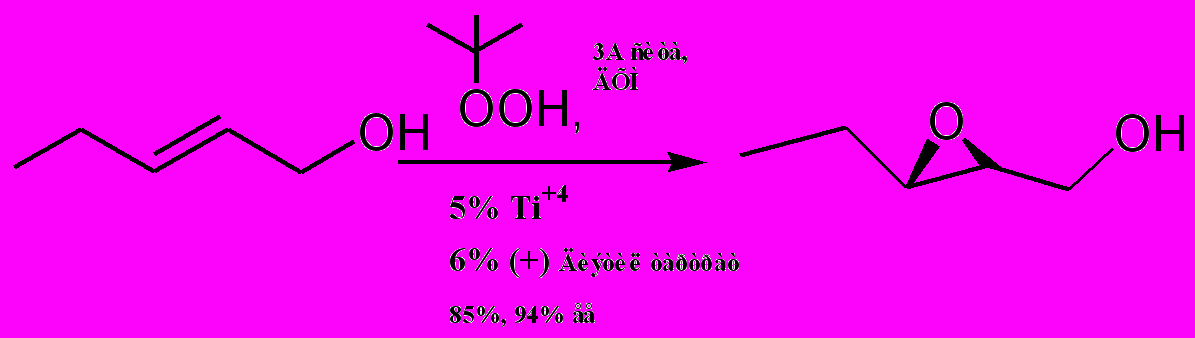
Схема 3
Американский химик Барри Шарплес (B.Sharpless) был удостоен Нобелевской премии в 2001 г. за развитие методов энантиоселективного эпоксидирования. (см. например M. G. Finn, K. B. Sharpless, J. Am. Chem. Soc. 113, 113 (1991) )
Электронодефицитная С=С связь устойчива к электрофильной атаке, но образует оксираны под действием нуклеофильных окислителе ( -OOH)
Альтернативным методом синтеза оксиранов является присоединение карбеновых частиц * к карбонильным соединениям (Схема 4)
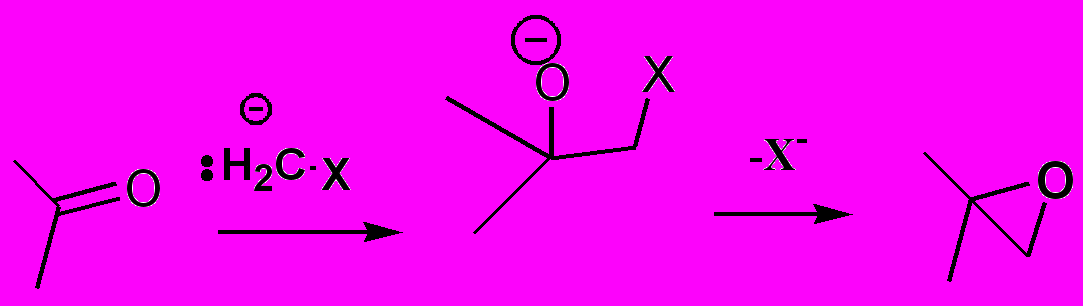
Схема 4
- - источником CH2 группы могут быть, например диазометан ( X= N2+), реагент Кори Me2S(O)=CH2 (X=OS+Me2) О методах синтеза реагента Кори см например (Synthetic Communic. 1985, 749)
Тииран
Один из удобных методов синтеза тииранов заключается в реакции 1,2-дизамещенного алкена смесью бис(триметилсилил)сульфида с бромом в безводном дихлорметаном при –78 С. Синтез является стереоспецифичным и селективным, т.к. в реакцию вступают только 1,2 дизамещенные алкены и стереохимия алкена сохраняется в тииране. (см. например Tetrahedron Lett., 1985, 4177) Схема 5
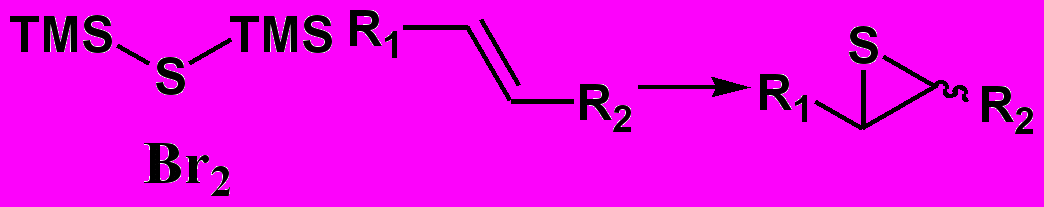
Схема 5
Также описан синтез тииранов присоединением диазоалкенов к тиокарбонильным соединениям. Катализатором этого процесса может быть, например CuCl. (Схема 6)
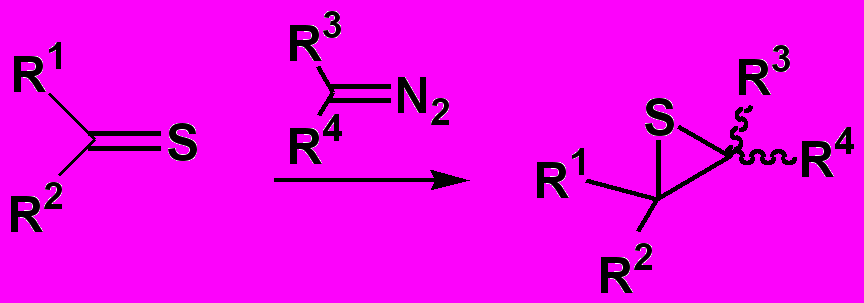
Схема 6
Лекция 3
Пиррол. Методы синтеза, реакционная способность
“ ^ Классические методы синтеза”
Синтез Пааля-Кнорра.
Исходным соединением в синтезе являются 1,4-дикарбонильные соединения или их циклические эквиваленты (например 2,5-диметоксифуран). (Схема 1)
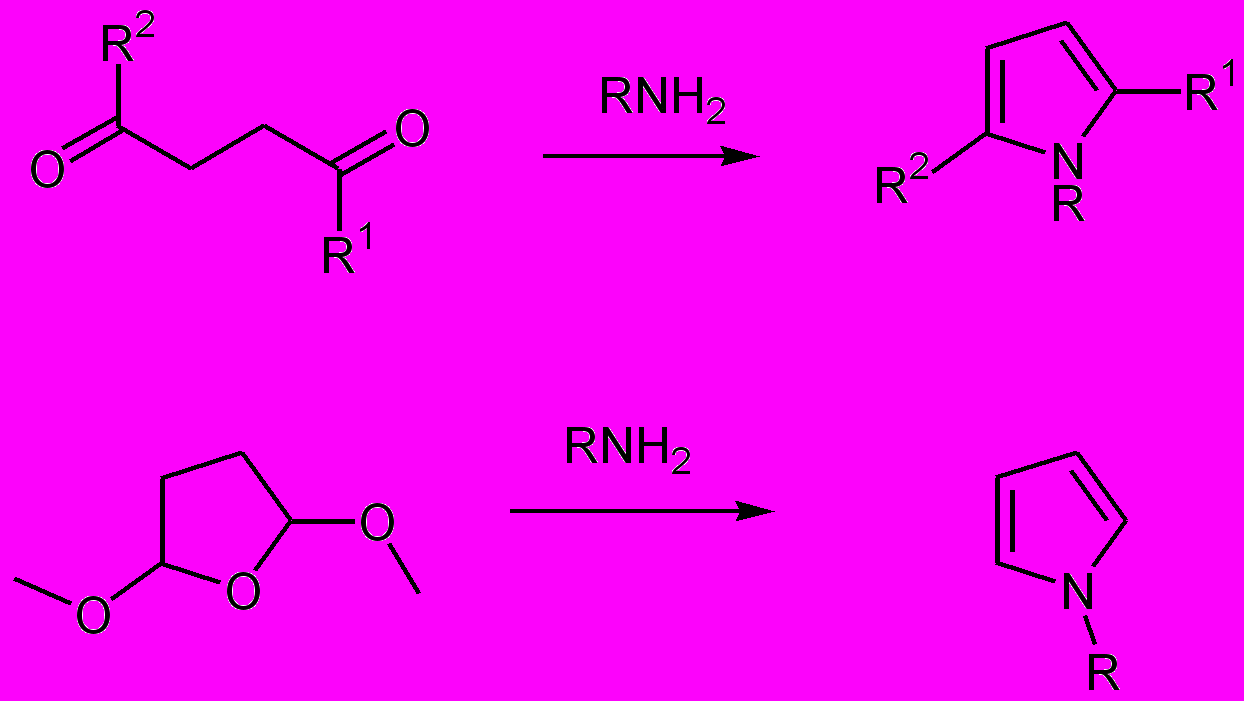
Схема 1
При использовании 2,5-диметоксифурана возможно введение арильных заместителей (использование аминов с низкими нуклеофильными свойствами)
^
Реакция Кнорра
В реакции участвуют две молекулы ацетоуксусного эфира. Из одной нитрозированием и восстановлением in situ получают аминокетон, который далее реагирует со второй молекулой. Использование бензилового или трет-бутилового эфира вместо этилового, облегчает последующее удаление алкоксикарбонильных групп. (Схема 2)
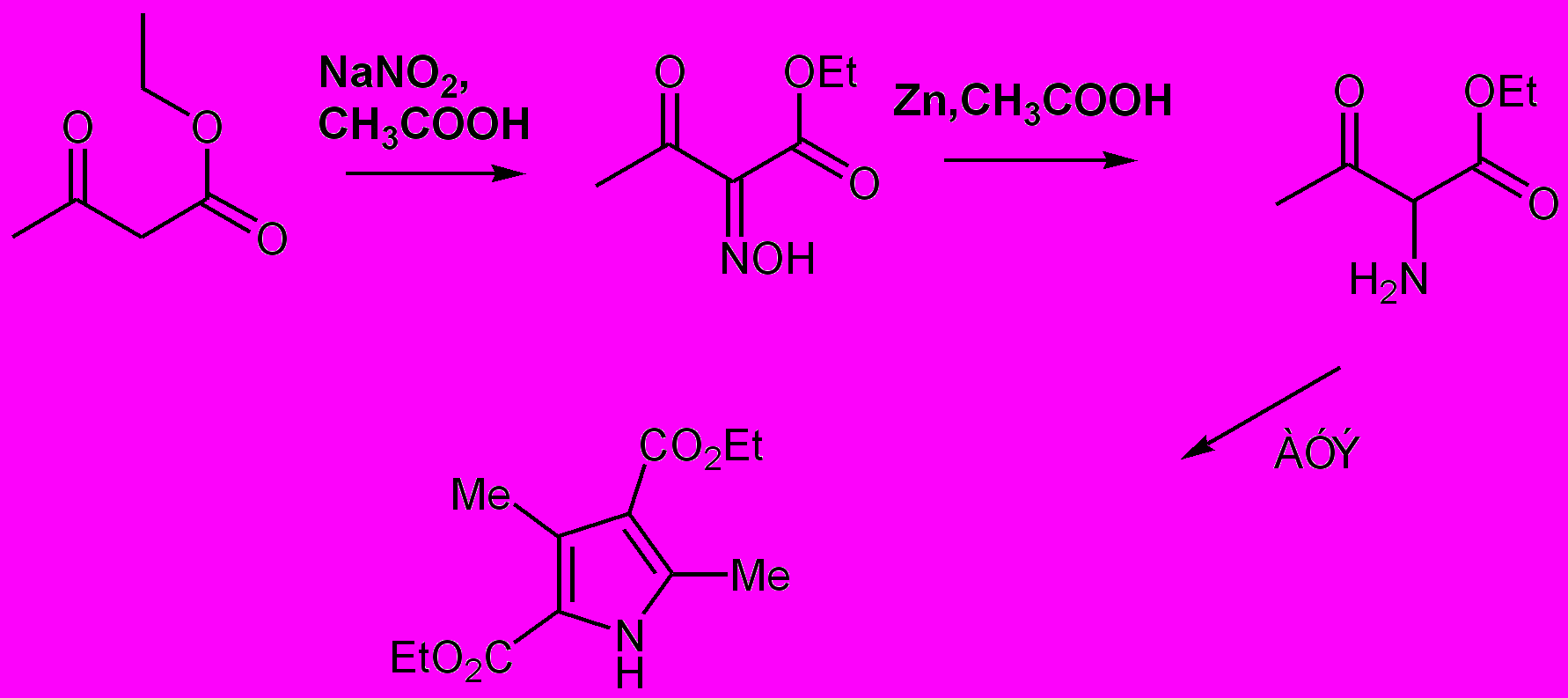
Схема 2
Интересной модификацией реакции Кнорра является синтез, предложенный профессором Mukaiayama ( см. например Angew.Chem. 1990, 777) . По этому методу можно получать 2,3-дизамещенные пирролы. (Схема 3)
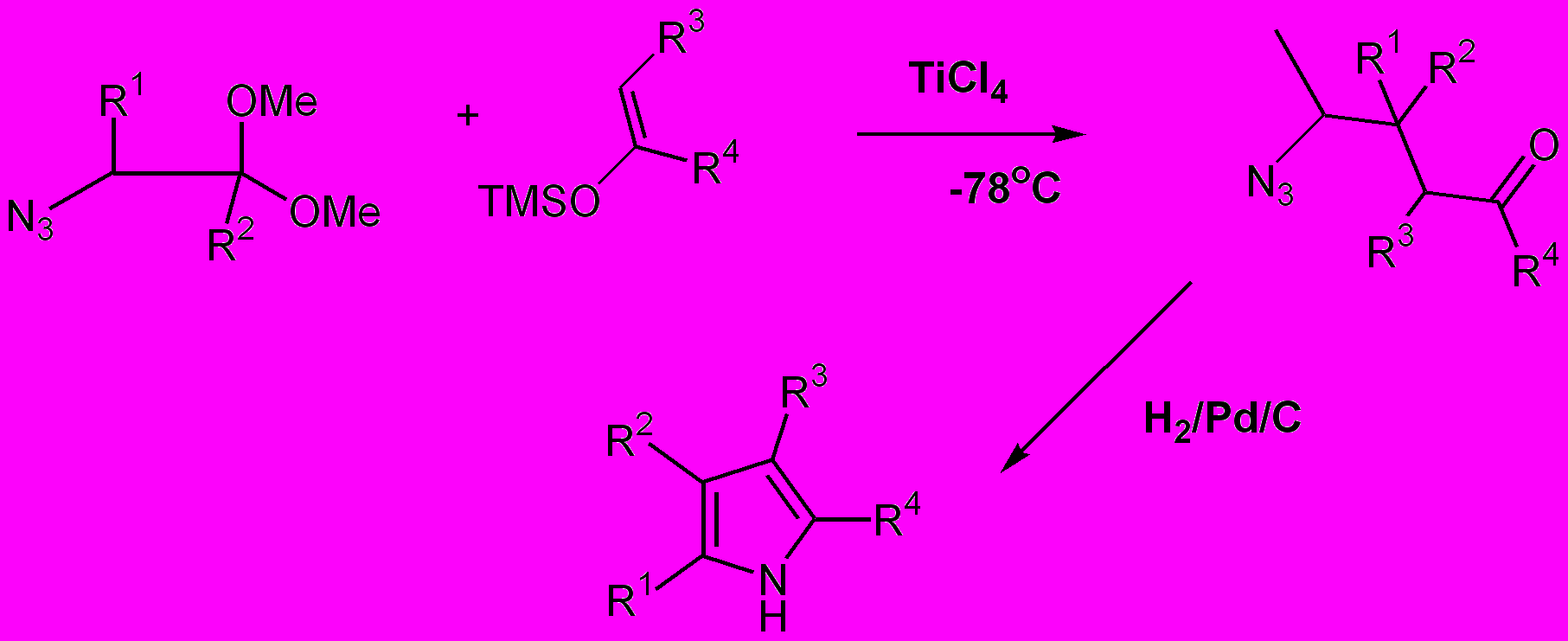
Схема 3
^
Синтез Ганча
Пирролы получают реакцией -галогенкетона с -кетоэфиром и аммиаком (Схема 4)
Метод применяется для получения некоторых эфиров 2,5-диалкилпиррол-3-карбоновых кислот.
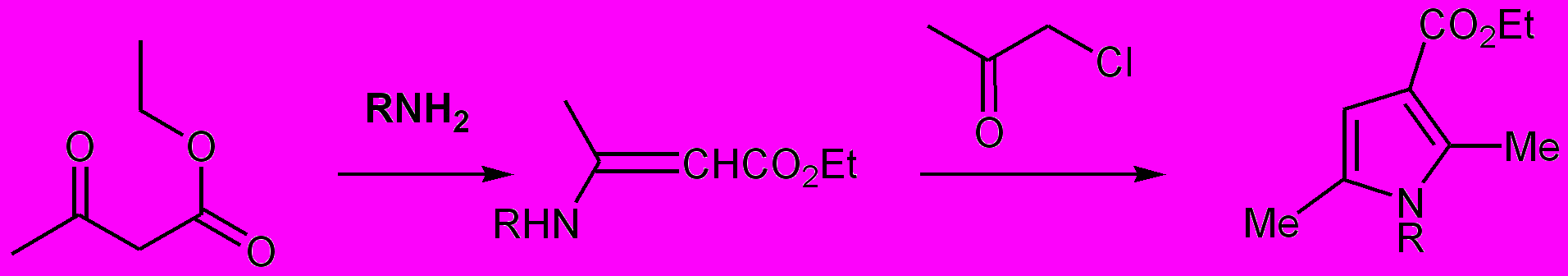
Схема 4
^
Другие методы синтеза
С высоким выходом замещенные пирролы образуются при взаимодействии азлактонов с ацетиленами. Азлактоны -гетероциклические соединения, образующиеся при взаимодействии аминокислот с хлорангидридами карбоновых кислот. Замыкание азлактонного цикла представляет, по сути, модификацию, приводящую одновременно к увеличению кислотности С-Н связи углерода, связанного с атомом азота. Вследствие этого азлактон способен вступать в реакцию присоединения по Михаэлю по ацетиленовой кратной связи. Далее следует замыкание цикла по связи C=N в составе азлактонного цикла и отщепление молекулы CO2, завершающее образование пиррольного цикла. (Схема 5)

Схема 5
^ Реакция Трофимова
Реакция гетероциклизации оксимов кетонов с ацетиленом открытая Б.А. Трофимовым в середине 70-х годов прошлого века, представляет собой удобный метод получения различно замещенных и аннелированных пирролов. Конденсация кетоксимов с ацетиленом требует применения суперосновных сред, в качестве которых используют гидроксиды щелочных металлов или тетраалкиламмония в диметилсульфоксиде. (Реакция может сопровождаться винилированием образующихся пирролов). Экспериментальные данные последних лет свидетельствуют в пользу [3,3]-сигматропного механизма. (Схема 6)
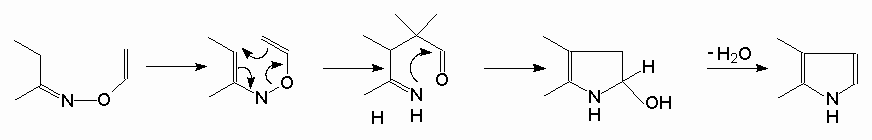
Схема 6
Пиррол в промышленности получают фракционной перегонкой каменноугольной смолы и костяного масла – вещества, образующегося при сухой перегонке костей. Кроме того, его получают из фурана по методу Юрьева, а также взаимодействием ацетилена с формальдегидом и аммиаком.
Сукцинимид, по сути, является производным пиррола и может быть превращен в пиррол восстановлением цинком.

^ Химические свойства пиррола
Пиррол представляет собой слабую NH-кислоту (рКа=17,5). Величина рКа значительно уменьшается при введении в ядро акцепторных заместителей. Синтетическое применение NH-кислотности пиррола заключается в N-металлировании и использовании металических производнях для контролируемых реакций электрофильного замещения. Регионаправленность замещения зависит от степени ковалентности связи азот-металл и от способности растворителя сольватировать катионы металла. Соли Na и К - ионные соединения, а Li и Mg - ковалентные, если в среде не присутствует диполярный апротонный растворитель (типа ГМФТА).
Для ковалентных солей алкилирование идет по атому углерода: (Схема 7)
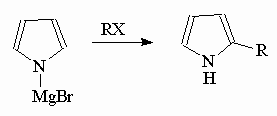
Для ионных солей алкилирование и ацилирование идет по атому азота при действии на пиррол, например, t-BuOK и использовании 18-краун-6 в качестве катализатора: (Схема 8)
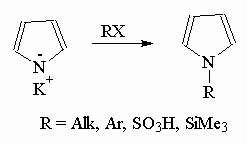
Схема 8
^ Реакции электрофильного замещения.
Говоря о реакциях электрофильного замещения в случае производных пиррола, необходимо помнить о его ацидофобности и , соответственно, невозможности использования кислот-содержащих реагентов. В кислых средах образующийся при протонировании катион электрофильно атакует следующую молекулу пиррола, вновь образующийся катион снова атакует непротонированную молекулу и т.д. В конечном итоге такой процесс приводит к полимеризации. (Схема 9)
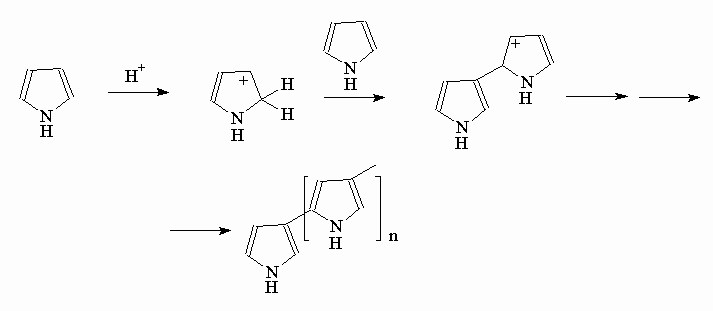
^ Схема 9
Нитрование
Обычно для нитрования пиррол-содержащих систем используют ацетил нитрат. При этом , если пиррол не замещен по атому азота, мажорным продуктом является 2-нитропроизводное, наличие алкильного заместителя приводит к образованию смеси 2- и 3-нитро производных, 3-нитропиррол можно селективно получить нитрованием N-фенилсульфонилпиррола с последующим снятием защитной группы. (См. например Tetrahedron Lett. 1981, p.4899 (Схема 10). В литературе имеются упоминания об ипсо-нитровании замещенных пирролов, сопровождающемся замещением на NO2- группу алкильных и ацильных радикалов.
Сульфирование
Пиррол сульфируется в положение 2 действием пиридин сульфотриоксида.
Галоидирование.
Пиррол легко галоидируется. Взаимодействием с эквимолярным количеством сульфурил хлорида в эфире при 0oC получают 2-хлоропиррол, дальнейшее хлорирование приводит к образованию ди-, три- и тетрахлорпроизводные. Реакция N-замещенных пирролов с N-бромсукцинимидом в ТГФ приводит к селективному образованию 2-бромпиррола. Бромирование и иодирование действием брома и иода в уксусной кислоте приводит к образованию тетрагалоген-производных.
Лекция 4
Фуран, тиофен – методы синтеза и реакционная способность
Фуран, тиофена и их производные (также как и рассмотренный ранее пиррол) могут быть получены из одних и тех же предшественников – 1,4-дикарбонильных соединенийпо реакции Пааля-Кнорра. Так, производные фурана образуются при действии на 1,4-дикетоны дегидратирующими веществами – (Р2О5, H2SО4, ZnCl2 и др.).
Реакция с неорганическими сульфидами, например с P2S5 приводит к образованию производных тиофена. (Схема 1)



Схема 1
1,4- дикарбонильные соединения могут быть использованы и в защищенной форме например в види диацеталя (Схема 2). ( см. J.Org.Chem. 1954, 1671)
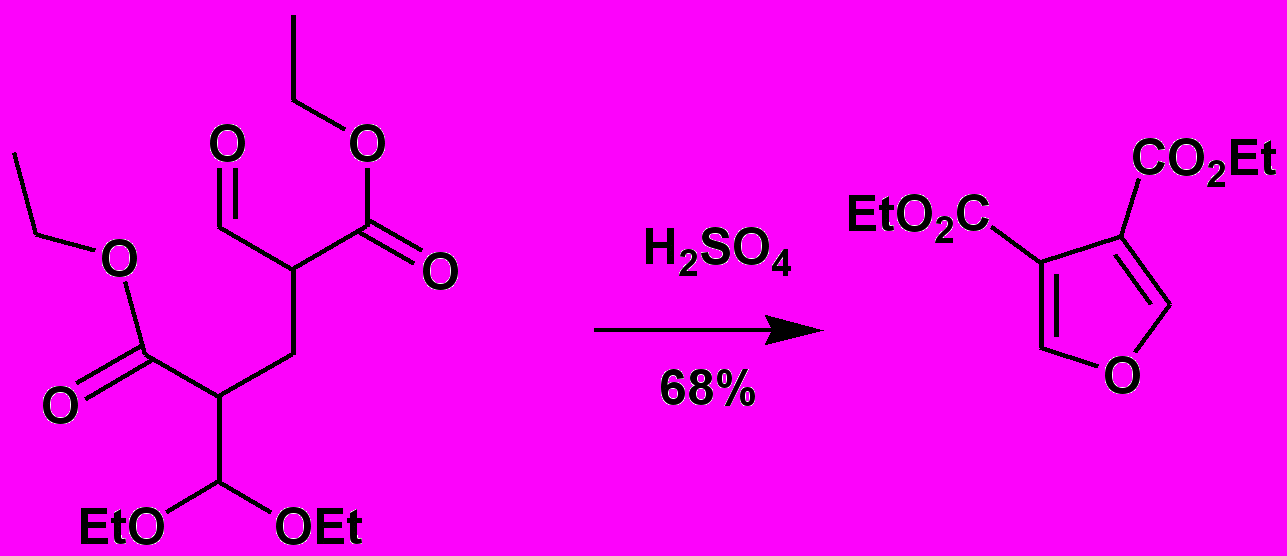
Схема 2
Поскольку образование гетероцикла пиррола, фурана и тиофена может происходить из одних и тех же исходных 1,4-дикарбонильных соединений, в соответствующих условиях возможны и их взаимопревращения. Эта реакция была открыта Юрьевым и носит его имя. Превращения происходят при нагревании гетероцикла в присутствие окиси алюминия при 400 °С в токе H2S, NH3 или H2O, однако высокий выход достигается только в случае использования фурана в качестве исходного соединения.
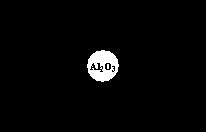 Схема 3
Схема 3Еще один общий метод получения пятичленных гетероциклов основан на использовании в качестве исходного соединения слизевой кислоты и других дикарбоновых кислот – продуктов окисления сахаров. Сухая перегонка слизевой кислоты приводит к образованию пирослизевой или a-фуранкарбоновой кислоты. При пиролизе аммонийной соли слизевой кислоты образуется пиррол.

Схема 4
Декарбоксилирование пирослизевой кислоты является методом получения фурана. Промышленным способом получения фурфурола – фуран-2-карбальдегида является кислотный гидролиз полисахаридов, в состав которых входят пентозы – пятиатомные сахара, содержащиеся в различном растительном сырье. Так, фурфурол получают из шелухи семян подсолнечника, кукурузных початков, соломы, отрубей, вследствие чего он и получил свое название (латинское furfur – отруби).

Схема 5
Далее фурфурол превращают в пирослизевую кислоту либо по реакции Канницаро, либо окислением кислородом воздуха в присутствии щелочных растворов солей меди или серебра, Полученную кислоту декарбоксилируют в фуран нагреванием до 200-250° С. Сам фурфурол так же может быть превращен в фуран нагреванием при 400 °С в присутствии катализаторов – хромитов цинка или марганца.
Тиофен обычно содержится в качестве примеси в бензоле, получаемом из каменноугольной смолы (до 0.5%). Температуры кипения бензола и тиофена близки (80 и 84 °С соответственно), что затрудняет их разделение перегонкой, однако бензол может быть очищен от примеси тиофена химически.
^
Задание на дом: Предложите химический метод отделения примеси тиофена от бензола, используя представления об относительной реакционной способности этих соединений.
В промышленности тиофен получают взаимодействием бутана, бутена или бутадиена с серой при высокой температуре (600°) с малым временем контакта реагирующих веществ – около 1 сек и немедленным охлаждением. При этом в реакцию вступает только часть реагентов, их отделяют от циклических продуктов, и вновь вводят в реакцию. Отметим, что сера в этой реакции выступает в качестве дегидрирующего окислителя, под действием которого гидрированные производные тиофена превращаются в тиофен.

Схема 6
Тиофен может быть получен в лабораторных условиях взаимодействием динатриевой соли янтарной кислоты с P2S3. Этот метод интересен тем, что и замещенные янтарные кислоты в тех же условиях образуют соответствующие замещенные производные тиофена.
Еще одно превращение, при котором образуется тиофен, открытое Чичибабиным, заключается во взаимодействии ацетилена с сероводородом при 400-450 °С на окиси алюминия. Эта реакция не имеет препаративного значения, однако, интересна своим сходством с реакций, тримеризации ацетилена с образованием бензола.
Схема 7
Интересным и эффективным методом синтеза 2-амино тиофенов является реакция Гевальда - внутримолекулярная циклизация непредельных нитрилов с участием CN группы и атома серы в качестве нуклеофила (схема )

Схема 8
^
Задача на дом – предложить механизм выделенной реакции
Химические свойства тиофена
Протонирование и реакции с ним связанные
При обработке тиофена горячей фосфорной кислотой происходит протонирование по -положению кольца и дальнейшая тримеризация .

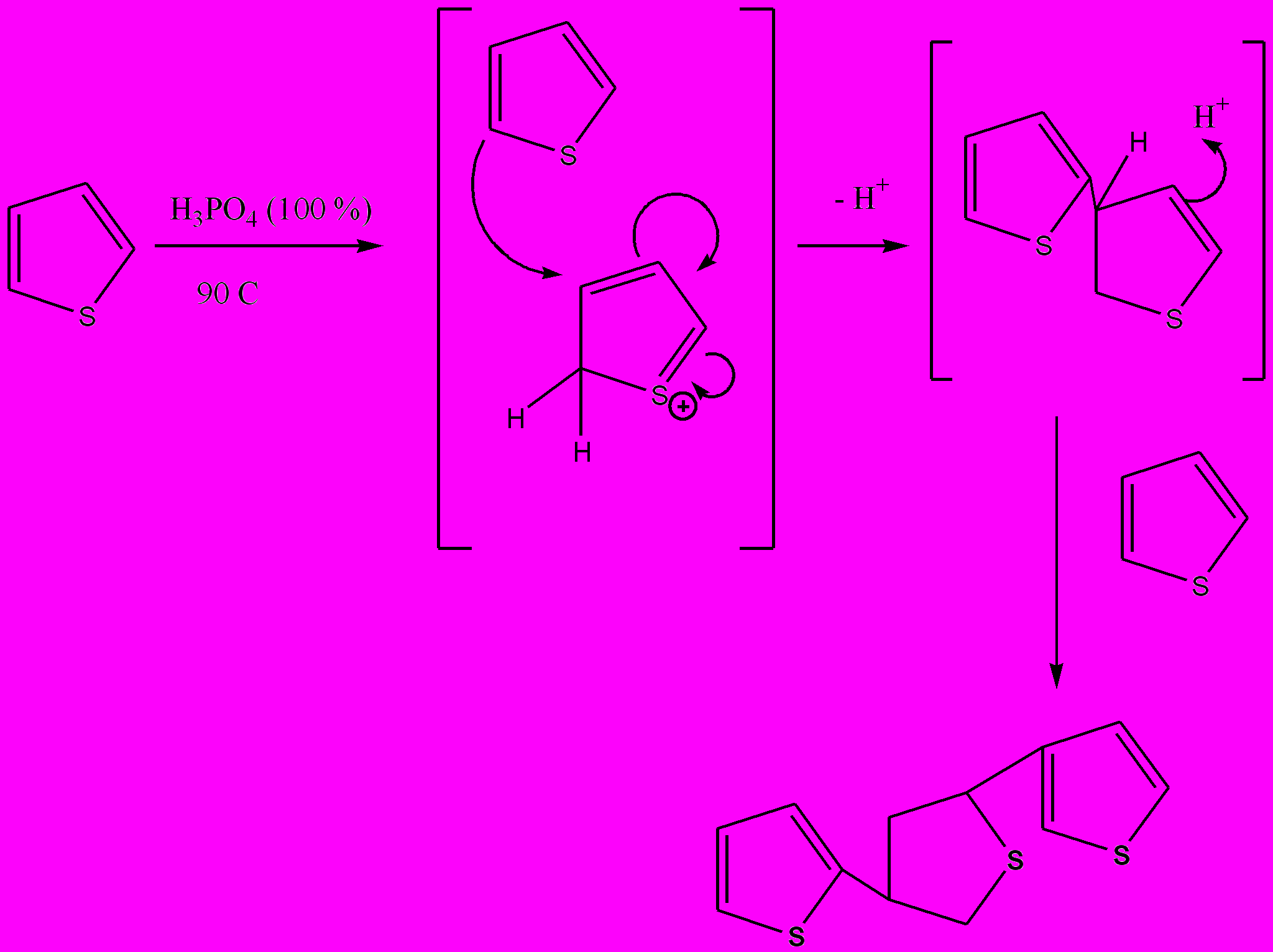
Схема 9
Нитрование
Для нитрования тиофена лучшим реагентом следует считать ацетилнитрат и тетрафторборат нитрония . Преимущественно получается 2-нитротиофен (6:1)
Сульфирование
Описано получение тиофен-2-сульфокислоты при сульфировании концентрированной серной кислотой, однако пиридин-сульфотриоксид является более удобным реагентом для этого синтеза.
2-хлорсульфонирование тиофена также представляет собой удобный препаративный метод.
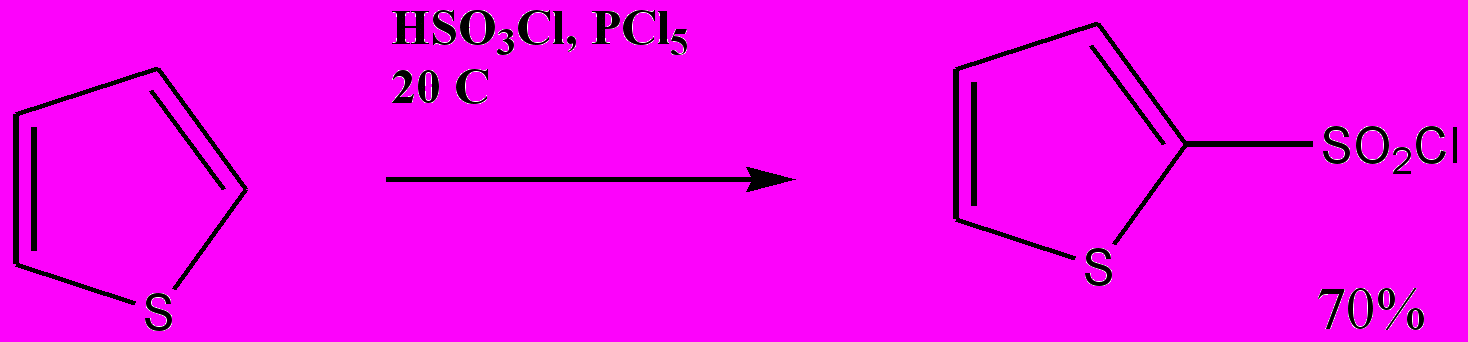
Схема 10
Ацилирование
Ацилирование по Фриделю-Крафтсу проходит достаточно хорошо, однако использование хлорида алюминия в качестве катализатора требует контролируемых условий проведения реакции (для того, чтобы избежать осмоления, необходимо катализатор добавлять к тиофену и ацилирующему реагенту, а не наоборот) . Предпочтительно использовать хлорид олова.
Формилирование по Вильсмайеру-Хааку протекает легко и приводит к селективному образованию 2-формилтиофена.
^ Конденсации с альдегидами и кетонами.
Легко протекает реакция хлорметилирования, катализируемая кислотой
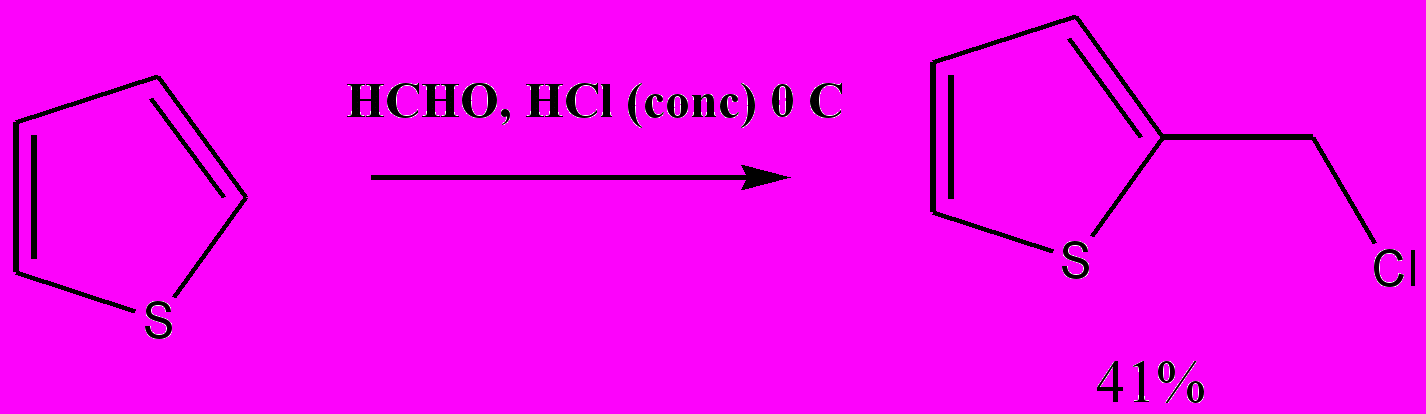
Схема 11
Гидроксиалкилирование 2-формилтиофена по положению 5 происходит при взаимодействии тиофенальдегида с другим альдегидом в присутствии иодида самария (II)
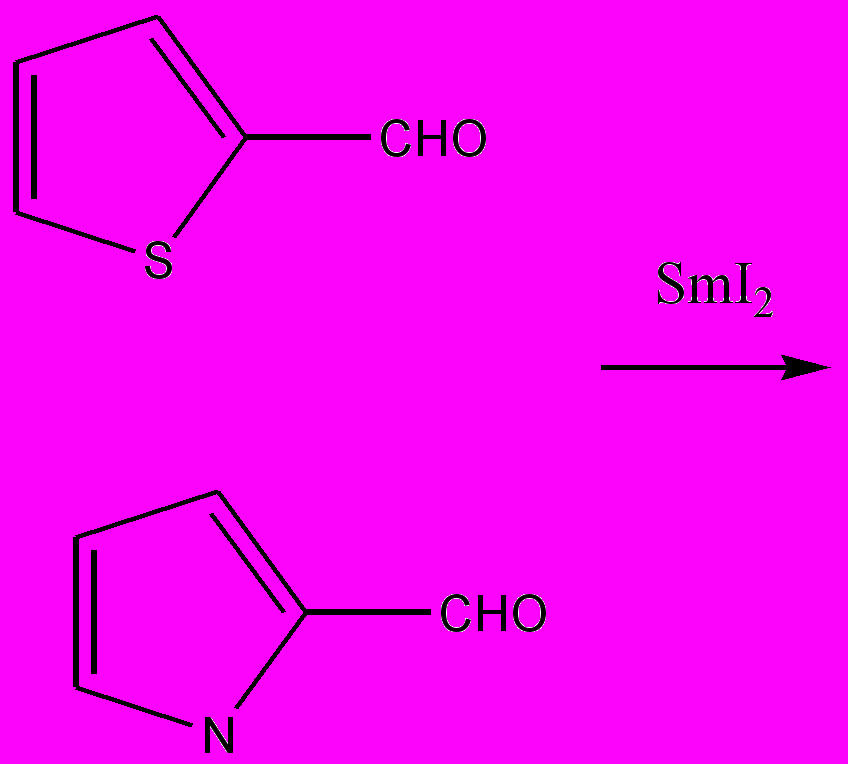
Схема 11
^
Лекции 5,6 Индол и его производные
Методы синтеза индола и его производных
Молекула индола содержит пиррольный гетероцикл, аннелированный с бензольным кольцом. Методы синтеза индола обычно основаны на замыкании пиррольного цикла в молекуле, содержащей бензольное кольцо. Примером такой реакции является взаимодействие анилина с ацетиленом при высокой температуре (Чичибабин). Эта реакция не имеет препаративного значения, а лишь демонстрирует сходство с реакцией получения пиррола взаимодействием аммиака с ацетиленом . (Схема 1)

Схема 1
Наиболее общий способ получения индола и его производных это синтез Фишера, заключающийся в перегруппировке фенилгидразонов альдегидов и кетонов в присутствии кислотных катализаторов – хлористого цинка, трехфтористого бора, полифосфорной кислоты и др. Наиболее вероятный механизм этой реакции включает орто-бензидиновую перегруппировку, которая протекает как синхронное внутримолекулярное превращение. (Схема 2)

Схема 2
^
Определите, на какой стадии (или стадиях) реакции требуется кислотный катализатор.
Предскажите преимущественное направление реакции в случае фенилгидразона метилэтилкетона.
Многие замещенные индолы и сам индол могут быть получены по методу Маделунга, заключающемуся в циклизации орто-ацетотолуидинов при действии сильных оснований. Выходы простых производных индола при этом бывают очень высокими. (Схема 3)

Схема 3
Механизм реакции Маделунга точно не известен, возможно, он и не один. Одно из наиболее простых объяснений протекания этой реакции строится на аналогии с конденсацией Кляйзена. Напишите схему этой реакции.
^Реакция Бишлера
Этот метод основан на кислотно-катализируемой циклизации -ариламино кетонов, альдегидов, либо их ацеталей. (Схема 4) (См. например JOC, 1981, p.778)
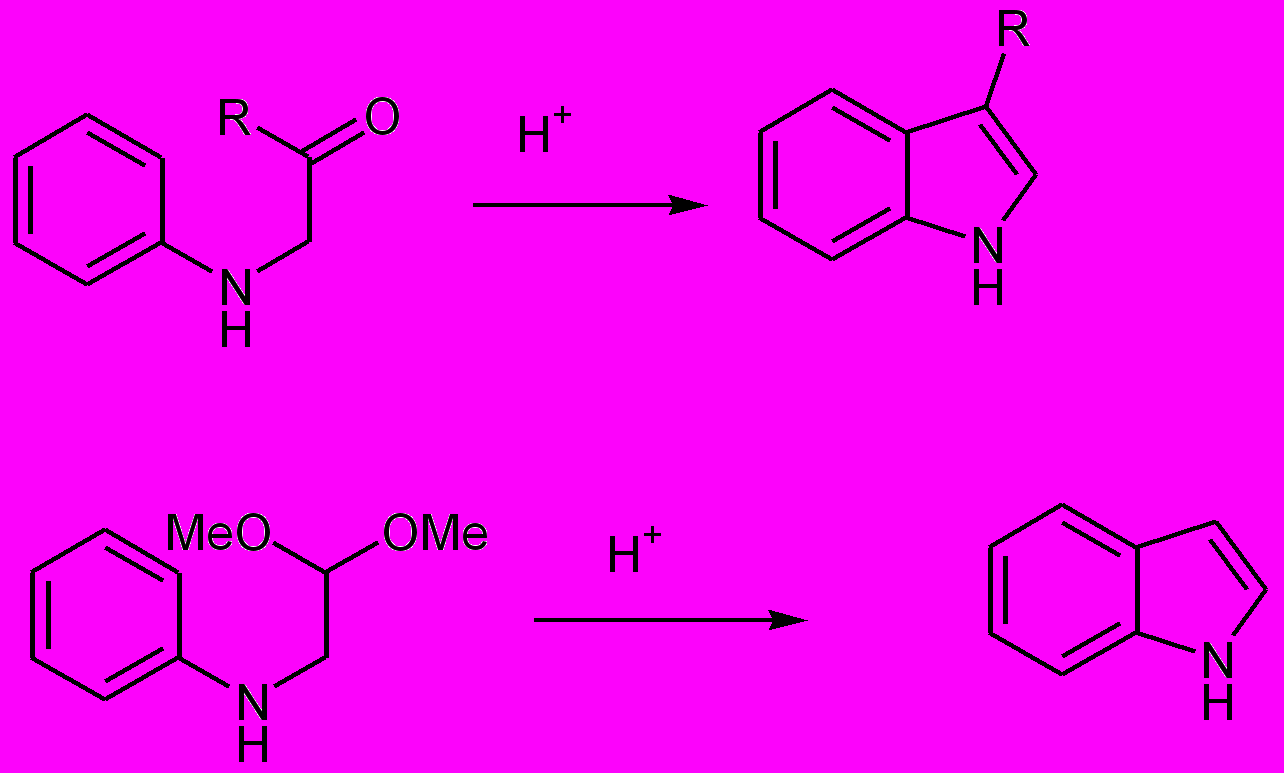
Схема 4
^
Внутримолекулярная реакция Хека (Heck).
Представляет собой относительно новый (см. JOC ,1980, p. 2709, JCS (Perkin trans. 1), 1993, p.1941) метод построения индольного ядра. Позволяет синтезировать индолы замещенные как по атому азота, так и по кольцу. Метод основан на катализируемой солями палладия внутримолекулярной циклизации N-аллил-o-галогенанилинов. (Схема 5)
^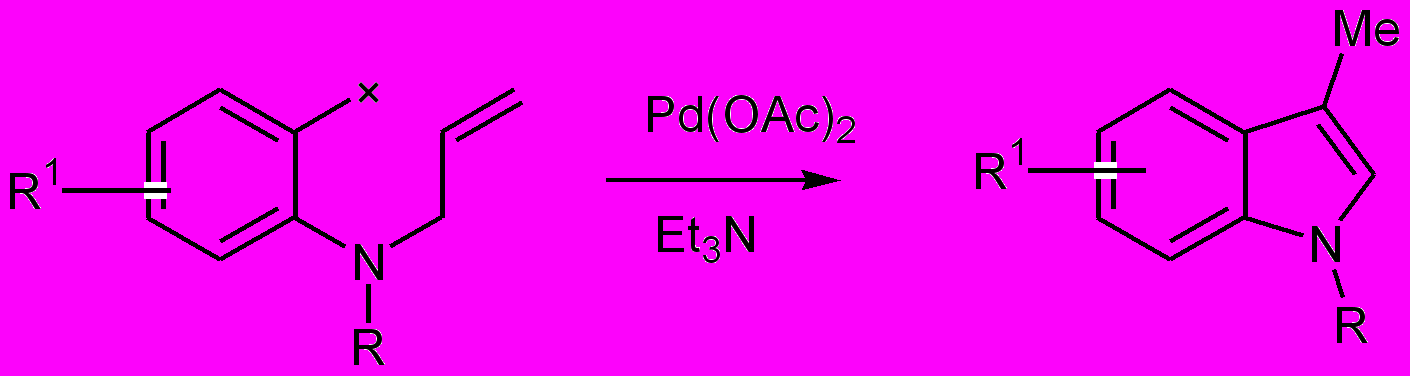
Схема 5
Синтез производных индола
Изатин
Изатины получают внутримолекулярным ацилированием производных анилинов (Схема 6) 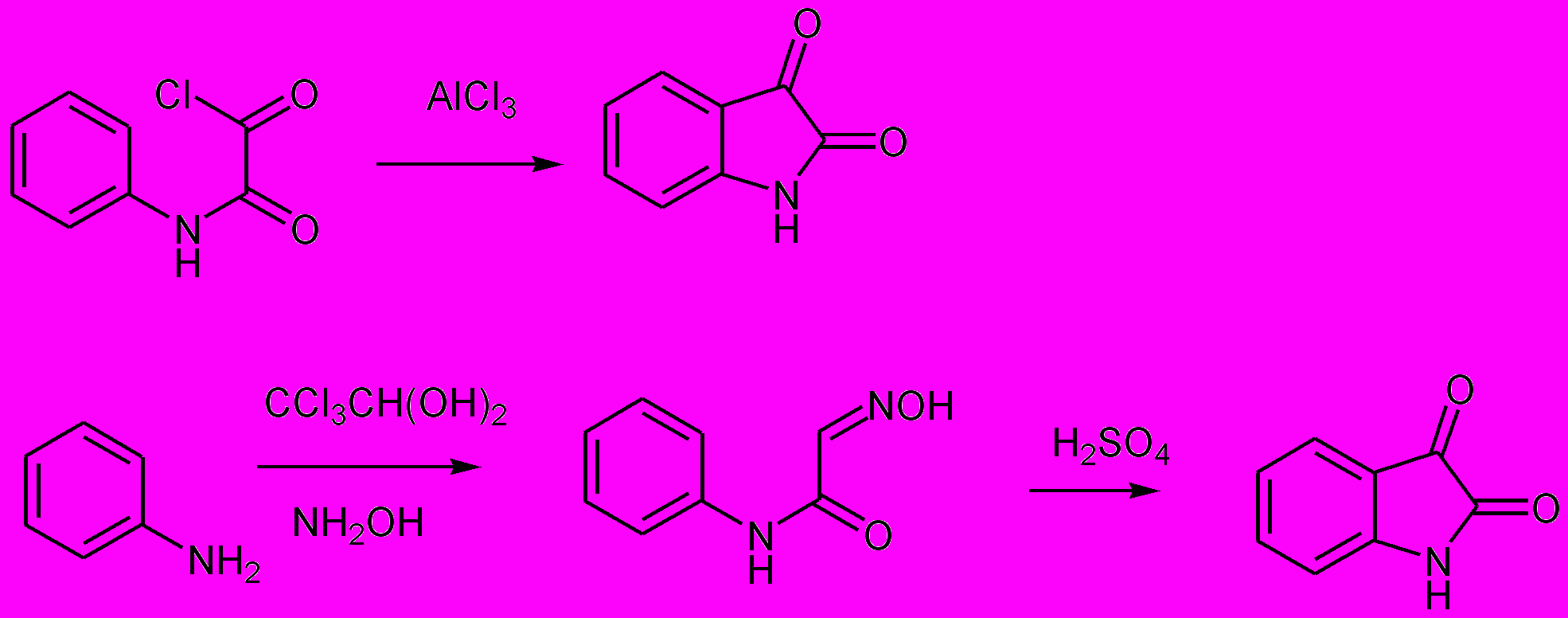
^
Свойства индола.
Индол, подобно пирролу, является ароматическим соединением, относящимся к p-избыточным гетероциклам. Это означает, что атом азота в составе гетероцикла увеличивает электронную плотность на атомах углерода как в пиррольной, так и, до некоторой степени, в бензольной части молекулы. Это приводит к тому, что в положении 3 возникает избыточная электронная плотность.
^
Изобразите резонансные структуры, демонстрирующие делокализацию пары электронов атома азота по p-системе индола
Учитывая вышесказанное, можно сделать вывод, что основность индола по атому азота, как и в случае пиррола, также должна быть пониженной, а реакционная способность в реакциях электрофильного замещения – повышенной, хотя и уступает реакционной способности пиррола (почему?). Индол, подобно пирролу, мало устойчив к действию кислот, поэтому реакции с электрофилами проводят, избегая сильнокислых сред. Преимущественное направление электрофильной атаки для индола – положение 3 гетероцикла, что находится в соответствии с эффективной резонансной стабилизацией образующегося при этом s-комплекса. Отметим, что в случае атаки по положению 2 резонансная стабилизация s-комплекса с участием гетероатома возможна только через делокализацию положительного заряда по бензольному кольцу, что менее выгодно вследствие нарушения ароматичности всей системы.
Примерами реакций индола, протекающих как электрофильное замещение, являются реакция Вильсмаейра, приводящая к 3-формилиндолу, и аминометилирование – реакция Maннuxa – взаимодействие с формальдегидом и солями вторичных аминов. При взаимодействии формальдегида с вторичным амином в кислой среде образуется соль N,N-диметилметилидениминия (изобразите механизм образования этой соли и объясните причину, по которой она является электрофилом), которая и выступает в роли электрофила. Надо отметить, что реакция Манниха характерна не только для индола и других -избыточных гетероциклов, но и для многих СН-активных соединений, например кетонов. В случае индола реакция с формальдегидом и хлоргидратом диметиламина приводит к грамину – алкалоиду, встречающемуся в некоторых растениях. Более важным является то обстоятельство, что грамин является исходным соединением в промышленном синтезе триптофана – незаменимой аминокислоты. С этой целью грамин вводят во взаимодействие с натриевой солью эфира ацетамидомалоновой кислоты, образующийся малоновый эфир гидролизуют и декарбоксилируют. Подробнее об этом можно прочесть в пособии, посвященном аминокислотам.

Еще одно полезное вещество, которое получают по реакции электрофильного замещения в индоле, это индолилуксусная кислота или гетероауксин – стимулятор роста растений. Гетероауксин образуется при взаимодействии индола с Na-солью хлоруксусной кислоты в щелочной среде и последующем подкислении реакционной смеси для выделения свободной кислоты. Это реакция алкилирования, то есть электрофильного замещения в ароматическом ядре, однако условия ее протекания принципиально отличаются от условий, характерных для реакции Фриделя-Крафтса. Возможность ее протекания связана с тем, что индол, подобно пирролу, является достаточно сильной NH-кислотой, способной депротонироваться с образованием аниона. Последний, будучи анионом, гораздо легче реагирует с электрофилом, чем исходный индол, и электрофильная атака направлена в положение 3.

s-комплекс
Приведенный выше пример показывает, что в реакции электрофильного замещения зачастую вводят не сам индол, а его анион, который обычно генерируют действием сильного основания, например магнийорганического соединения. В отличие от аниона пиррола в этом случае реакции протекают в несколько более жестких условиях и всегда по положению 3. Еще один пример такого рода превращения это взаимодействие индола с амилнитритом в присутствии метилата натрия, которое приводит к оксиму.

Следует отметить, что во всех случаях электрофильное замещение в индоле происходит по положению 3. Если положение 3 занято электронодонорным заместителем, то реакция протекает по положению 2, и только если в положении 3 находится электроноакцепторный заместитель, реакция может идти по бензольной части молекулы. Это означает, что если нужно синтезировать индол, замещенный в бензольном кольце, необходимо чтобы заместитель уже присутствовал на стадии построения гетероцикла. Например, 4-оксизамещенные индолы могут быть получены взаимодействием циклогександиона-1,3 с a-бромкетонами и последующей реакцией с первичными аминами или аммиаком.
 | К производным 4-оксииндола относится псилоцибин, обладающий галлюциногенным действием. В природе это соединение встречается в некоторых грибах, которые шаманы использовали для погружения в состояние транса. Среди 5-оксипроизводных индола также встречаются вещества, обладающие сильным влиянием на высшую нервную деятельность. Среди них серотонин, нарушение концентрации которого в организме, ведет к шизофрении. Сложное вещество – диэтиламид лизергиновой кислоты (ЛСД), в молекуле которого также содержится индольный фрагмент, при попадании в организм нарушает баланс серотонина и приводит к очень ярким цветным галлюцинациям. ЛСД является самым сильным галлюциногеном из всех известных веществ, галлюцинации вызываются приемом нескольких микрограммов этого препарата. Получают его из лизергиновой кислоты, которая содержится в спорынье – грибке, паразитирующем на зерновых, главным образом, на ржи. С давних времен известны случаи отравления людей, употреблявших в пищу зерно, зараженное спорыньей. |
Лекции 7,8
^ Хинолин, изохинолин
Методы получения хинолина и изохинолина
Так же, как и пиридин, хинолин и изохинолин содержатся в каменноугольной смоле, откуда они могут быть выделены благодаря тому, что, подобно пиридину, являются основаниями.

Основным методом получения хинолина является синтез Скраупа, заключающийся во взаимодействии анилина или замещенных анилинов с глицерином, в присутствии серной кислоты и окислителя, в качестве которого часто используют нитробензол, мета-нитробензолсульфокислоту, а иногда и просто кислород воздуха.
На первой стадии глицерин под действием серной кислоты дегидратируется с образованием акролеина. Далее происходит присоединение анилина к акролеину по типу реакции Михаэля. Вследствие протонирования альдегидной группы в образующемся интермедиате происходит ее активация как электрофила, приводящая к замыканию гетероцикла в результате электрофильного замещения по орто-положению бензольного кольца. Последующая дегидратация приводит к дигидропроизводному хинолина, которое окисляется имеющимся в реакционной среде окислителем в хинолин. Используемый в качестве окислителя нитроарен восстанавливается в соответствующий анилин. (Схема 1)



Схема 1
В реакцию Скраупа могут быть вовлечены и ненасыщенные кетоны. Модификацией метода Скраупа является синтез Дебнера-Миллера. В этом случае в реакцию с анилином вводят алифатические альдегиды в присутствии кислот. При этом на первой стадии происходит кротоновая конденсация альдегида с образованием ненасыщенного альдегида, который далее реагирует с анилином по схеме, приведенной выше. При таком варианте проведения реакции нет необходимости в использовании дополнительного окислителя, роль которого в этом случае выполняет исходный альдегид. (Схема 2)

Схема 2
Удобным методом синтеза производных хинолина является взаимодействие ариламинов с дикетонами и последующая циклизация промежуточно образующихся енаминокетонов при действии серной кислоты.
^ Синтез Комба - конденсация ариламинов с 1,3-дикарбонильными соединениями
На первом этапе конструирования пиридинового цикла молекулы хинолина в этом синтезе происходит образование основания Шиффа, дальнейшая электрофильная циклизация которого идет, вероятно, через дипротонированный интермедиат. Дегидратация промежуточной 4-гидрокси-1,4-дигидроструктуры приводит к ароматизации. (Схема 3)


80%
(Джоуль и Смит, стр. 119-120)
Схема 3
^ Катализируемые палладием конденсации о-галогенанилинов с аллиловыми спиртами. Синтез хинолонов-4 из о-галогенанилинов, ацетиленов и СО.
В качестве исходных соединений для синтеза хинолинов могут служить о-галогензамещенные анилины. Для образования связи С(4)-С(4а) в этом случае требуется применение палладиевых катализаторов. (Tetrah.Lett. - 1991. - Vol. 32. - P. 569)
Например, о-иоданилины конденсируются с замещенными аллиловыми спиртами, образуя 2-замещенные хинолины. Процесс идет по схеме: (Схема 4)


Схема 4
Наиболее часто используемый метод синтеза изохинолинов носит название реакции Бишлера-Напиральского. Схема синтеза заключается в циклизации -фенилэтиламидов под действием кислотного катализатора, в качестве которого используют фосфорный ангидрид с хлорокисью фосфора или полифосфорную кислоту. Обычно образующийся дигидроизохинолин превращают в изохинолин каталитическим дегидрированием – нагреванием в присутствии платинового катализатора. (Схема 5)

Схема 5
Циклизация по Бишлеру-Напиральскому, как и стадия циклизации в синтезе Скраупа, является ароматическим электрофильным замещением, в котором электрофилом является атом углерода протонированной карбонильной группы. Этим определяется характер влияния заместителя, находящегося в бензольном кольце, на скорость реакции, ее направление и выход целевого продукта. При взаимодействии мета-хлоранилина с глицерином в присутствии серной кислоты и мета-нитробензолсульфокислоты в качестве окислителя образуется примерно одинаковое количество 5-хлор- и 7-хлорхинолинов. В то же время, конденсация по Бишлеру-Напиральскому мета-метоксифенилацетамида приводит исключительно к 6-метоксипроизводному (выход 50%), 8-метоксипроизводное не образуется. (Схема 6)


Схема 6
Протеканию циклизации по Бишлеру-Напиральскому не препятствует и введение в бензольное кольцо электроноакцепторной нитрогруппы, правда выход продукта циклизации при этом заметно снижается (< 10%). Следует отметить, что и не содержащий заместителя в бензольном кольце фенилацетамид циклизуется в соответствующее дигидропроизводное с выходом всего 23%. Низкий выход производных изохинолина компенсируется доступностью исходных соединений.
Большое значение в синтезе алкалоидов, являющихся производными изохинолина, имеет метод Пикте-Шпенглера, в котором в циклизацию вводят не ацетамидные производные, как в методе Бишлера-Напиральского, а имины, образующиеся при взаимодействии альдегидов с фенилэтиламинами. В том случае, когда в бензольном кольце присутствуют активирующие заместители, реакция циклизации происходит в очень мягких, иногда даже в физиологических условиях (рН среды, температура, концентрации реагентов), и приводит к гидрированным производным изохинолина с высоким выходом. ( Схема 7)

Химические свойства хинолинов и изохинолинов
Химические свойства хинолинов и изохинолинов имеют много общего со свойствами пиридинов, однако существуют некоторые особенности, обусловленные наличием аннелированного пиридинового ядра.
Атмы азота в хинолине (рКа=4,94) и изохинолине (рКа=5,40) обладают основными свойствами. Аналогично пиридину они легко протонируются, кватернизуются, образуют комплексы с кислотами Льюиса (BF3, SO3 и т.п.).
Электрофильное замещение в хинолинах и изохинолинах идет только по бензольному кольцу в положения 5 и 8, все реакции идут в катионах хинолиния и изохинолиния.
| Электрофил | Реагенты и условия | Основные продукты |
| Хинолин | ||
| D+ | D2SO4 (70%), 150oC | 8 |
| NO2+ | HNO3, H2SO4, 0oC | 5 и 8 (1:1) |
| Br+ | Br2, AlCl3, 80oC | 5*1 |
| SO3H+ | H2SO4, SO3, 90oC | 8*2 |
| Изохинолин | ||
| D+ | D2SO4 (90%), 180oC | 5 |
| NO2+ | HNO3, H2SO4, 0oC | 5 и 8 (9:1) |
| Br+ | Br2, AlCl3, 75oC | 5*3 (78%) |
*1 Образуется некоторое количество 8-бромхинолина; при избытке брома получают 86% 5,8-дибромхинлина.
*2 При 220оС образуется 5-хинолинсульфокислота, при 300оС 5- и 8-сульфопроизводные перегруппировываются в термодинамически более стабильный 6-изомер.
*3 При использовании 2 моль брома образуется 5,8-дибромизохинолин.
Есть несколько исключений, когда электрофильные заместители вступают в пиридиновое кольцо хинолина и изохинолина (однако эти реакции идут с очень низкими выходами):

При нагревании гидрохлоридов хинолина и изохинолина с бромом в нитробензоле также образуются продукты замещения по пиридиновому фрагменту - 3-бромхинолин и 4-бромизохинолин соответственно:

Атомы азота в пиридиновом фрагменте молекул этих гетероциклов активируют их к нуклеофильному замещению, которое для хинолина идет в положения 2 и 4, а для изохинолина - в положение 1. Нуклеофильное замещение в положение 3 изохинолина невыгодно, т.к. делокализация отрицательного заряда с участием гетероатома приводит к нарушению ароматичности системы:

Нуклеофильное замещение для хинолина и изохинолина идет по механизму присоединения-элиминирования. Замещение атома хлора на алкокси-, фенилсульфо-, амино- и другие группы (в том числе карбанионы) можно проводить селективно с учетом различной способности атомов хлора к нуклеофильному замещению в различных положениях ядра:

Замещение гидрид-иона идет аналогично реакции Чичибабина в пиридиновом ряду.

Аминирование 2-незамещенных хинолинов амидом натрия в диметиланилине идет с низкими выходами, существенно лучшие результаты дает использование амида бария в жидком аммиаке. Аминирование 2-замещенных хинолинов идет по положению 4 существенно легче, чем в пиридине.
Гидроксилирование хинолина сухим КОН в жестких условиях приводит к получению хинолона-2, использование для этих целей гипохлорита натрия протекает существенно легче, т.к. в промежуточно образующемся катионе N-хлорхинолиния облегчается нуклеофильное замещение.

Основным отличием хинолинов и изохинолинов от пиридинов является их склонность к реакциям по кольцу, содержащему гетероатом. Координация нуклеофила по атому азота ведет к присоединению нуклеофила по соседнему положению:

Так, в случае хинолина гидроксид N-метилпиридиния существует в равновесии с продуктом 1,2-присоединения - псевдооснованием:

Для хинолина и изохинолина известно образование ковалентных аддуктов - продуктов присоединения цианид-иона к N-ацилпиридиниевым солям - соединений Рейссерта, которые можно использовать в синтетических целях, например, для введения заместителя в положение 1 изохинолина:

Восстановление хинолинов и изохинолинов алюмогидридом лития приводит к нестойким 1,2-дигидроструктурам, которые легко диспропорцианируются. При использовании в качестве восстановителя олова в соляной кислоте или при каталитическом гидрировании образуются устойчивые 1,2,3,4-тетрагидрохинолины и изохинолины.
Окисление хинолинов и изохинолинов перманганатом калия в щелочной среде приводит, как правило, к разрушению бензольного кольца и образованию пиридиндикарбоновых кислот. Однако, в зависимости от строения соединений может быть окислено и пиридиновое ядро.
Образующаяся при окислении хинолина дикрбоновая кислота декарбоксилируется с образованием никотиновой кислоты. При окислении изохинолина в тех же условиях соответствующая дикарбоновая кислота образуется в смеси с ее ангидридом.

Окисление хинолина пероксидом водорода или надкислотами приводит, к образованию N-оксидов, в которых, также как и в ряду пиридина, облегчается электрофильное замещение и изменяется его региоориентация. Так, N-оксид хинолина легко нитруется в положение 4. Как известно, N-оксидная группировка способствует и нуклеофильному замещению, что дает возможность легко замещать, например, ввденную электрофильно нитрогруппу на различные нгуклеофилы:

Интересным свойством N-оксида хинолина является их способность при облучении УФ светом претерпевать различные перегруппировки. Одним из направлений при этом служит образование хинолона-2, которое идет через промежуточное образование трехчленного цикла, его электроциклическое раскрытие и миграцию протона. Другим вариантом превращения трициклического интермедиата является [1,5]-сигматропная перегруппировка в изомерный трицикл, электроциклическое раскрытие которого ведет к расширению пиридинового кольца в термодинамически более стабильный семичленный цикл:


^ Лекция 9 Диазины
Методы синтеза шестичленных азотистых гетероциклов с двумя гетероатомами (диазинов).
К шестичленным азотистым гетероциклам с двумя гетероатомами относятся пиридазин, пиримидин и пиразин.

Из этих трех гетероциклических систем наибольшее значение имеет пиримидин, поэтому о синтезе и свойствах пиридазина и пиразина будут даны только краткие сведения. Фрагменты пиримидина, а также пурина (см. ниже) входят в состав жизненно важных нуклеиновых кислот.
Наиболее широко используемый метод синтеза производных пиридазина заключается во взаимодействии 1,4-дикетонов с гидразином. Ненасыщенные 1,4-дикетоны, также используемые для построения пиридазинового цикла, могут быть получены гидролитическим расщеплением фуранов. Кроме того, пиридазиновое кольцо формируется в результате реакции Дильса-Альдера между бутадиеном и эфирами азодикарбоновой кислоты.

Производные пиразина образуются в результате самоконденсации -аминокетонов. Эта реакция идет настолько легко, что -аминокетоны с незамещенной аминогруппой часто могут существовать только в виде солей, тогда как свободные основания немедленно вступают в реакцию конденсации. Образующиеся при этом производные дигидропиразина могут быть превращены в пиразины действием мягких окислителей, например солей двухвалентной ртути. Сам пиразин получают окислением 2,5-диметилпиразина в пиразиндикарбоновую кислоту и последующим ее декарбоксилированием.

Полностью гидрированное производное пиразина – пиперазин синтезируют взаимодействием дибромэтана с аммиаком. Отметим, что пиперазин образуется и при каталитическом гидрировании пиразина.
Существует три группы методов синтеза производных пиримидина. Первый из них основан на реакции конденсации соединений, в состав которых входит цепочка из трех атомов углерода и концевые атомы углерода способны реагировать с азотистым нуклеофилом, содержащими фрагмент N-C-N. В качестве первой компоненты используются -дикарбонильные соединения – -диальдегиды, -кетоальдегиды, -кетоэфиры, малоновые эфиры и тому подобное. Азотистой компонентой реакции может быть мочевина, тиомочевина, амидины, гуанидин. К этому типу реакций относится взаимодействие малоновых эфиров с мочевиной, приводящее к производным барбитуровой кислоты. При взаимодействии замещенных малоновых эфиров с мочевиной также образуются производные барбитуровой кислоты – барбитураты, которые используются в качестве снотворных препаратов. Реакция ,-диэтилмалонового эфира с мочевиной приводит к соединению, которое известно как «барбитал» или «веронал», – первому снотворному из ряда барбитуратов, нашедшему практическое применение в терапии. Изобразите структуру этого соединения.



Другими примерами синтеза производных пиримидина по этой методологии является взаимодействие гуанидина с 4,4-диэтокси-2-бутаноном – ацеталем -кетоальдегида. При этом образуется 2-амино-4-метилпиримидин с количественным выходом. Реакция малонодинитрила с тиомочевиной приводит к диаминомеркаптопиримидину. (Предложите механизм этой реакции.)
При взаимодействии натриевой соли формилуксусного эфира с мочевиной образуется урацил.
Предложите метод синтеза формилуксусного эфира.
Вторая группа методов построения пиримидинового кольца основана на взаимодействии -дикарбонильных соединений или их производных, содержащих "скрытую" карбонильную функцию, с формамидом. При этом образуются производные пиримидина, не имеющие заместителя в положении 2.

И, наконец, третья группа синтезов основана на взаимодействии соединений, содержащих карбонильную функцию и аминогруппу в -положении к ней, с соединениями, содержащими фрагмент C-N. Примером такой реакции является взаимодействие эфиров -аминопропионовой кислоты с изоцианатом калия. При этом образуется гидрированное производное пиримидина, которое окисляют в пиримидин действием брома в уксусной кислоте.

^ Свойства диазинов.
В составе гетероцикла диазинов присутствуют два электроноакцепторных атома азота, что настолько снижает реакционную способность этих соединений в реакциях электрофильного замещения, что провести их с незамещенными диазинами не удается. По той же причине диазины являются очень слабыми основаниями, образующими соли только при действии сильных кислот, причем протонированию подвергается лишь один из атомов азота. Алкилирование незамещенных диазинов также происходит только по одному из атомов азота, их окисление надкислотами приводит только к моно-N-оксидам. Введение в состав гетероцикла активирующих заместителей, таких как N-оксидная группа, гидрокси- или аминогруппа, позволяет проводить реакции электрофильного замещения. В случае пиримидина эти реакции идут по положению 5 – наименее дезактивированному электроноакцепторным влиянием гетероциклических атомов азота.
Объясните, почему положение 5 в пиримидиновом кольце является предпочтительным для электрофильной атаки.

Таким образом, реакции электрофильного замещения не характерны для азинов. Значительно чаще в синтезе производных азинов используются реакции нуклеофильного замещения. При этом наиболее активными положениями в гетероцикле являются и -положения относительно гетероатома. Определите, какие положения для различных азинов являются предпочтительными для нуклеофильной атаки. Обоснуйте свой выбор.
Представленные выше методы синтеза азинов приводят, как правило, к оксипроизводным, которые легко могут быть превращены в хлорпроизводные при действии пятихлористого фосфора. Таким образом, из барбитуровой кислоты может быть получен сначала трихлопиримидин, а затем и сам пиримидин. Восстановительное дехлорирование проводят йодистым водородом.

При гидролизе нуклеиновых кислот среди прочих соединений образуются три производных пиримидина – урацил, тимин и цитозин.

Цитозин может быть получен из урацила с использованием реакций нуклеофильного замещения. На первой стадии взаимодействием с пятихлористым фосфором урацил превращают в дихлорпиримидин, который далее вводят во взаимодействие с аммиаком. Реакция происходит неселективно и приводит к двум изомерным аминохлорпиримидинам, гидролиз которых приводит к цитозину и изоцитозину.

Другой метод синтеза цитозина основан на реакции конденсации натриевой соли формилуксусного эфира с S-этилтиомочевиной. В полученном производном 4-оксипиримидина проводят замещение оксигруппы на атом хлора, образующийся хлорпиримидин вводят во взаимодействие с аммиаком. В этом случае реакция нуклеофильного замещения происходит селективно по положению 4, поскольку хлорид-анион – существенно лучшая уходящая группа, чем этилтиолят-анион. Последняя стадия синтеза – гидролиз – приводит к цитозину.

Таким образом, используя последовательность реакций конденсации и нуклеофильного замещения, удается синтезировать различные функциональные производные пиримидина, в том числе и азотистые основания нуклеотидов.
Предложите метод синтеза тимина.
К производным пиримидина относится один из наиболее эффективных и распространенных сульфамидных антибиотиков сульфадимезин (предложите метод синтеза этого соединения). Пиримидиновый фрагмент входит в состав молекулы витамина B1 или тиамина, недостаток которого в организме вызывает нервное заболевание бери-бери.
