
Федеральная программа книгоиздания России Рецензенты: канд психол наук С. А. Исайчев, доктор биол наук И. И. Полетаева Равич-Щербо И. В. и др. Р12
| Вид материала | Программа |
- Вестник балтийской педагогической академии вып. 94. – 2010 г. Актуальные проблемы нравственного, 2431.92kb.
- Рецензенты: профессор, доктор психол наук Филонов Л. Б., вед науч сотрудник, канд психол, 2609.63kb.
- Научный выпуск вестник балтийской педагогической академии вып. 29. – 2000 г. Поиск, 1745.18kb.
- Общеобразовательная программа дошкольного образования Авторский коллектив, 5619.19kb.
- Образовательная программа дошкольного образования Москва «Просвещение», 5670.3kb.
- Введенским Игорем Витальевичем Рецензенты доктор психол наук В. А. Лабунская канд психол, 375.9kb.
- Отчет о проведении Международной научной конференции-семинара «Современные методы психологии», 97.76kb.
- Приглашение и программа разнообразие почв и биоты северной и центральной азии, 521.14kb.
- Пояснительная записка, 12621.4kb.
- В организации совместных отношений, 1137.14kb.
D -мужчина; о -Женщина; П-О- мя рецессивными аллелями (хх). супружеская пара; l-g3 - супружеская Родитель, страдающий ХГ, чаще пара и их ребенок; ♦ или ■-носитель всег0 является носителем геноти-заболсвания; Л или М — пробонд - Па Хх и в момент скрещивания носитель заболевания, через которого порождает гамету (яйцо или спер-были собраны сведения о родословной. мий\ либо с х, либо с х аллелем. (Подробнее о правилах составления гс- г
Фактических Древ см. гл. VII.) Гаметы нормального родителя
всегда содержат рецессивные аллели х. Четыре возможных комбинации этих аллелей показаны на рис. 2.2. Дети таких родителей всегда наследуют один здоровый аллель, передаваемый по наследству нормальным родителем. Однако, поскольку при зачатии аллели родителей комбинируются по случайному закону, для каждого из потомков вероятность наследования аллеля X от родителя, страдающего ХГ,
Рис. 2.2. Схема скрещивания: аллельный механизм наследования хореи Гентингтона (пример доминантного наследования).
[Х] — доминантный аллель, вызывающий развитие ХГ; [х] — рецессивный аллель (здоровый).
72
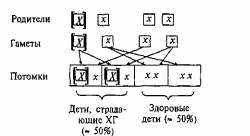
составляет 50%. Этим и объясняется тот факт, что у родителей, пораженных ХГ, только 50% детей страдают тем же заболеванием.
Для ХГ характерна одна особенность: первые симптомы этого заболевания проявляются лишь в зрелом возрасте, т.е. тогда, когда большинство людей уже создали семью и обзавелись детьми. В ином случае пациенты, страдающие ХГ, вообще не могли бы иметь потомков, так как умирали бы до наступления половой зрелости. Передача по наследству доминантного аллеля X возможна именно потому, что его летальный эффект не проявляется до начала репродуктивного периода.
Эта особенность развития ХГ создает чрезвычайно щепетильную психологическую ситуацию. В 1993 г. ученые открыли ген на хромосоме 4, вызывающий ХГ, и разработали молекулярно-генетический метод, позволяющий тестировать каждого человека с тем, чтобы определить, является ли данный индивидуум носителем патологического аллеля-мутанта (аллеля X).
Представьте себе следующую ситуацию. Ваши бабушка и дедушка по материнской линии умерли достаточно рано, и в семье не сохранилось никаких свидетельств того, что один из них, возможно, был носителем гена ХГ. Вашей матери 53, она больна ХГ. Вам 30, и у Вас есть возможность обратиться в лабораторию клинической генетики с тем, чтобы Вам сказали, являетесь Вы носителем гена X или нет. Вероятность того, что Вы — носитель этого гена, достаточно велика и составляет приблизительно 50%. Захотите ли Вы пройти подобный тест?
Исследования показывают, что большинство взрослых людей, для которых риск развития ХГ высок (поскольку один из родителей болен), предпочитают подобный тест не проходить. Этот тест, однако, имеет принципиально другое значение в пренатальной диагностике, когда заранее можно определить, является ли развивающийся организм носителем аллеля X. Ранняя пре-натальная диагностика позволяет родителям сделать осмысленный выбор относительно жизни их будущего ребенка, а также создает возможность раннего пренатального клинического вмешательства.
РЕЦЕССИВНОЕ НАСЛЕДОВАНИЕ: ФЕНИЛКЕТОНУРИЯ
Закон расщепления объясняет и наследование фенилкетонурии (ФКУ) — заболевания, развивающегося в результате избытка важной аминокислоты — фенилаланина (Phe) в организме человека. Избыток фенилаланина приводит к развитию умственной отсталости. Частота встречаемости ФКУ относительно низка (примерно 1 на 10 000 новорожденных), тем не менее около 1% умственно отсталых индивидуумов страдают ФКУ, составляя, таким образом, сравнительно большую группу пациентов, умственная отсталость которых объясняется однородным генетическим механизмом.
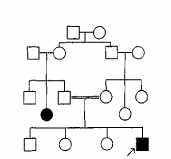
Как и в случае ХГ, исследователи изучали частоту встречаемости ФКУ в семьях пробандов. Оказалось, что пациенты, страдающие ФКУ, обычно имеют здоровых родителей. Кроме того, было замечено, что ФКУ чаще встречается в семьях, в которых родители являются кровными родственниками. Пример семьи пробанда, страдающего ФКУ,
73
оказан на рис. 2.3: больной ребенок родился у фенотипически здоровых родителей-кровных родственников (двоюродных брата и сестры), но сестра отца ребенка страдает ФКУ.
ФКУ передается по рецессивному типу наследования, т.е. генотип больного содержит два аллеля ФКУ, полученные от обоих родителей. Потомки,
Рис. 2.3. Пример родословной семьи, в которой ФКУ передается по наследству (тетя пробанда страдает этим заболеванием).
Двойная линия между супругами обозначает кровнородственный брак. Остальные
обозначения те же, что и на рис. 2. 1
которые имеют только один такой аллель, не страдают заболеванием, но являются носителями аллеля ФКУ и могут передать его своим детям. На рис. 2.4 показаны пути наследования аллелей ФКУ от двух фенотипически нормальных
родителей. Каждый из родителей имеет один аллель ФКУ и один нормальный аллель. Вероятность того, что каждый ребенок может унаследовать аллель ФКУ от каждого из родителей, составляет 50%. Вероятность того, что ребенок унаследует аллели ФКУ от обоих родителей одновременно, составляет 25% (0,5 х 0,5 = 0,25; вероятности умножаются, поскольку события наследования аллелей от каждого из родителей независимы друг от друга). Ген ФКУ и его структурные варианты, встречающиеся в разных популяциях, хорошо изучены. Знания, имеющиеся в нашем распоря-
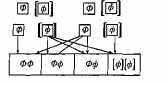
Родители
Гаметы
Потомки
Рис. 2.4. Схема скрещивания: аллельный механизм наследования ФКУ.
Ф — доминантный аллель («здоровый»); [ф]—рецессивный аллель, вызывающий развитие заболевания. ФФ, Фф — фенотипически нормальные дети (их 75%); только 25% имеют нормальный генотип (ФФ); еще 50% фенотипически здоровы, но являются носителями аллеля ФКУ (Фф). Оставшиеся 25% потомков — больны (1Ф11Ф1)-
74
жении, позволяют проводить своевременную пренатальную диагностику с тем, чтобы определить, унаследовал ли развивающийся зародыш две копии аллеля ФКУ от обоих родителей (факт такого наследования резко повышает вероятность заболевания). В некоторых странах, например в Италии, где частота встречаемости ФКУ достаточно высока, такая диагностика проводится в обязательном порядке для каждой беременной женщины.
Как уже отмечалось, ФКУ чаще встречается среди тех, кто вступает в брак с кровными родственниками. Несмотря на то что встречаемость ФКУ сравнительно низка, примерно 1 человек из 50 является носителем аллеля ФКУ. Вероятность того, что один носитель аллеля ФКУ вступит в брак с другим носителем такого аллеля, составляет примерно 2%. Однако при заключении брака между кровными родственниками (т.е. если супруги принадлежат к одной родословной, в которой аллель ФКУ передается по наследству) вероятность того, что оба супруга окажутся носителями аллеля ФКУ и одновременно передадут два аллеля будущему ребенку, станет значительно выше 2%.
4. ЗАКОН НЕЗАВИСИМОГО КОМБИНИРОВАНИЯ (НАСЛЕДОВАНИЯ) ПРИЗНАКОВ (ТРЕТИЙ ЗАКОН МЕНДЕЛЯ)
Этот закон говорит о том, что каждая пара альтернативных признаков ведет себя в ряду поколений независимо друг от друга, в результате чего среди потомков первого поколения (т.е. в поколении F2) в определенном соотношении появляются особи с новыми (по сравнению с родительскими) комбинациями признаков. Например, в случае полного доминирования при скрещивании исходных форм, различающихся по двум признакам, в следующем поколении (F2) выявляются особи с четырьмя фенотипами в соотношении 9:3:3:1. При этом два фенотипа имеют «родительские» сочетания признаков, а оставшиеся два - новые. Данный закон основан на независимом поведении (расщеплении) нескольких пар гомологичных хромосом. Так, при дигибридном скрещивании это приводит к образованию у гибридов первого поколения (F1) 4 типов гамет (АВ, Ав, аВ, ав), а после образования зигот - к закономерному расщеплению по генотипу и, соответственно, по фенотипу в следующем поколении (F2).
Парадоксально, но в современной науке огромное внимание уделяется не столько самому третьему закону Менделя в его исходной формулировке, сколько исключениям из него. Закон независимого комбинирования не соблюдается в том случае, если гены, контролирующие изучаемые признаки, сцеплены, т.е. располагаются по соседству друг с другом на одной и той же хромосоме и передаются по наследству как связанная пара элементов, а не как отдельные элементы. Научная интуиция Менделя подсказала ему, какие признаки дол-
75
жны быть выбраны для его дигибридных экспериментов, — он выбрал несцепленные признаки. Если бы он случайно выбрал признаки, контролируемые сцепленными генами, то его результаты были бы иными, поскольку сцепленные признаки наследуются не независимо друг от друга.
С чем же связана важность исключений из закона Менделя о независимом комбинировании? Дело в том, что именно эти исключения позволяют определять хромосомные координаты генов (так называемый локус*).
В случаях когда наследуемость определенной пары генов не подчиняется третьему закону Менделя, вероятнее всего эти гены наследуются вместе и, следовательно, располагаются на хромосе в непосредственной близости друг от друга. Зависимое наследование генов называется сцеплением, а статистический метод, используемый для анализа такого наследования, называется методом сцепления. Однако при определенных условиях закономерности наследования сцепленных генов нарушаются. Основная причина этих нарушений - явление крос-синговера, приводящего к перекомбинации (рекомбинации) генов. Биологическая основа рекомбинации заключается в том, что в процессе образования гамет гомологичные хромосомы, прежде чем разъединиться, обмениваются своими участками (подробнее о рекомбинации — в гл. I и IV).
Кроссинговер - процесс вероятностный, а вероятность того, произойдет или не произойдет разрыв хромосомы на данном конкретном участке, определяется рядом факторов, в частности физическим расстоянием между двумя локусами одной и той же хромосомы. Кроссинговер может произойти и между соседними локусами, однако его вероятность значительно меньше вероятности разрыва (приводящего к обмену участками) между локусами с большим расстоянием между ними.
Данная закономерность используется при составлении генетических карт хромосом (картировании). Расстояние между двумя локусами оценивается путем подсчета количества рекомбинаций на 100 гамет. Это расстояние считается единицей измерения длины гена и называется сентиморганом в честь генетика Т. Моргана, впервые описавшего группы сцепленных генов у плодовой мушки дрозофилы — любимого объекта генетиков. Если два локуса находятся на значительном расстоянии друг от друга, то разрыв между ними будет происходить так же часто, как при расположении этих локусов на разных хромосомах.
Используя закономерности реорганизации генетического матери-
* Напомним, что локусом (лат. locus - место) называется местоположение определенного гена или маркёра (полиморфного участка ДНК) на генетической карте хромосомы. Иногда термин «локус» неоправданно используют как синоним понятия «ген». Такое применение его неточно, поскольку речь может идти о положении не только гена, но и маркёра, находящегося в межгенном пространстве.
76
ала в процессе рекомбинации, ученые разработали статистический метод анализа, называемый анализом сцепления.
* * *
Законы Менделя в их классической форме действуют при наличии определенных условий. К ним относятся:
1) гомозиготность исходных скрещиваемых форм;
2) образование гамет гибридов всех возможных типов в равных соотношениях (обеспечивается правильным течением мейоза; одинаковой жизнеспособностью гамет всех типов; равной вероятностью встречи любых гамет при оплодотворении);
3) одинаковая жизнеспособность зигот всех типов.
Нарушение этих условий может приводить либо к отсутствию расщепления во втором поколении, либо к расщеплению в первом поколении; либо к искажению соотношения различных генотипов и фенотипов. Законы Менделя имеют универсальный характер для всех диплоидных организмов, размножающихся половым способом. В целом они справедливы для аутосомных генов с полной пенетрантностью (т.е. 100-процентной частотой проявления анализируемого признака; 100% пенетрантность подразумевает, что признак выражен у всех носителей аллеля, детерминирующего развитие этого признака) и постоянной экспрессивностью (т.е. постоянной степенью выраженности признака); постоянная экспрессивность подразумевает, что фенотипичес-кая выраженность признака одинакова или примерно одинакова у всех носителей аллеля, детерминирующего развитие этого признака.
Знание и применение законов Менделя имеет огромное значение в медико-генетическом консультировании и определении генотипа фенотипически «здоровых» людей, родственники которых страдали наследственными заболеваниями, а также в выяснении степени риска развития этих заболеваний у родственников больных.
Глава III
НЕМЕНДЕЛЕВСКАЯ ГЕНЕТИКА
Гениальность законов Менделя заключается в их простоте. Строгая и элегантная модель, построенная на основе этих законов, служила генетикам точкой отчета на протяжении многих лет. Однако в ходе дальнейших исследований выяснилось, что законам Менделя подчиняются только относительно немногие генетически контролируемые признаки. Оказалось, что у человека большинство и нормальных, и патологических признаков детерминируются иными генетическими
77
ала в процессе рекомбинации, ученые разработали статистический метод анализа, называемый анализом сцепления.
* * *
Законы Менделя в их классической форме действуют при наличии определенных условий. К ним относятся:
1) гомозиготность исходных скрещиваемых форм;
2) образование гамет гибридов всех возможных типов в равных соотношениях (обеспечивается правильным течением мейоза; одинаковой жизнеспособностью гамет всех типов; равной вероятностью встречи любых гамет при оплодотворении);
3) одинаковая жизнеспособность зигот всех типов.
Нарушение этих условий может приводить либо к отсутствию расщепления во втором поколении, либо к расщеплению в первом поколении; либо к искажению соотношения различных генотипов и фенотипов. Законы Менделя имеют универсальный характер для всех диплоидных организмов, размножающихся половым способом. В целом они справедливы для аутосомных генов с полной пенетрантностью (т.е. 100-процентной частотой проявления анализируемого признака; 100% пенетрантность подразумевает, что признак выражен у всех носителей аллеля, детерминирующего развитие этого признака) и постоянной экспрессивностью (т.е. постоянной степенью выраженности признака); постоянная экспрессивность подразумевает, что фенотипичес-кая выраженность признака одинакова или примерно одинакова у всех носителей аллеля, детерминирующего развитие этого признака.
Знание и применение законов Менделя имеет огромное значение в медико-генетическом консультировании и определении генотипа фенотипически «здоровых» людей, родственники которых страдали наследственными заболеваниями, а также в выяснении степени риска развития этих заболеваний у родственников больных.
Глава III
НЕМЕНДЕЛЕВСКАЯ ГЕНЕТИКА
Гениальность законов Менделя заключается в их простоте. Строгая и элегантная модель, построенная на основе этих законов, служила генетикам точкой отчета на протяжении многих лет. Однако в ходе дальнейших исследований выяснилось, что законам Менделя подчиняются только относительно немногие генетически контролируемые признаки. Оказалось, что у человека большинство и нормальных, и патологических признаков детерминируются иными генетическими
77
механизмами, которые стали обозначать термином «неменделевская генетика». Таких механизмов существует множество, но в этой главе мы рассмотрим лишь некоторые из них, обратившись к соответствующим примерам, а именно: хромосомные аберрации (синдром Дауна); наследование, сцепленное с полом (цветовая слепота); импринтинг (синдромы Прадера-Вилли, Энгельмана); появление новых мутации (развитие раковых заболеваний); экспансия (инсерция) повторяющихся нук-леотидных последовательностей (миотоническая дистрофия Дюшенна); наследование количественных признаков (сложные поведенческие характеристики).
1. ХРОМОСОМНЫЕ АБЕРРАЦИИ: СИНДРОМ ДАУНА
Синдром Дауна (СД) - одно из весьма ограниченного числа наследуемых заболеваний, фенотип которого хорошо известен даже неспециалистам. Его «известность» является результатом того, что, во-первых, частота встречаемости СД достаточно высока и, во-вторых, фенотип этого заболевания легко узнаваем: больным СД свойственны характерные внешние черты, выражение лица и умственная отсталость.
Первые клинические и научные описания СД появились в середине прошлого века, а его точное определение было дано в 1866 г. Дж. Дауном, описавшим несколько таких пациентов. Гипотезы о том, что СД контролируется генетически, были сформулированы в начале XX в. К 30-м годам было высказано предположение, что это заболевание развивается в результате аберрации хромосом (структурных отклонений в хромосомном наборе), причиной которой служит их нерасхождение в процессе мейоза. В 1959 г. было обнаружено, что СД вызывается трисомией хромосомы 21, т.е. наличием в клетках трех, а не двух, как обычно, хромосом. Сегодня известно, что примерно 1 из 600 новорожденных является носителем этой аномалии. Кроме того, по современным оценкам, примерно 1 из 150 оплодотворенных яйцеклеток человека является носительницей трисомии 21 (большинство яйцеклеток с трисомиями гибнет). Пациенты с СД составляют около 25% всех умственно отсталых, формируя самую большую этиологически однородную группу умственно отсталых.
Генетический механизм СД представляет собой иллюстрацию явления хромосомных аберраций. О них уже шла речь в гл. I. Коротко повторим сказанное там. Во время формирования половых клеток — гамет — все 23 пары хромосом делятся, и каждая гамета становится носителем одной хромосомы из каждой пары. Когда спермий оплодотворяет яйцеклетку, хромосомные пары восстанавливаются, причем в каждой паре одна хромосома приходит от матери, вторая — от отца. Несмотря на отлаженность процесса образования гамет, в нем случаются ошибки, и тогда разделение хромосомных пар нарушает-
78
ся — появляется гамета, которая содержит не одну хромосому, а их пару. Это нарушение называется нерасхождением хромосом. Когда такая гамета при оплодотворении сливается с нормальной гаметой, образуется клетка с тремя одинаковыми хромосомами; подобное явление и называется трисомией (см. рис. 1.7). Нерасхождение хромосом служит главной причиной спонтанных абортов в течение первых нескольких недель жизни плода. Тем не менее существует некоторая вероятность того, что зародыш с аномальным хромосомным набором продолжит развитие.
Точная причина нерасхождения неизвестна. Надежным коррелятом трисомии-21 является возраст матери: согласно исследованиям, у 56% матерей старше 35 лет плоды оказываются носителями трисомии-21, и в таких случаях примерно 90% диагностированных женщин предпочитают искусственно прервать беременность. Поскольку СД появляется «заново» в каждом поколении (нерасхождение — единичное событие, вероятность появления которого увеличивается с возрастом матери), постольку СД нельзя рассматривать как заболевание, передающееся по наследству.
2. НАСЛЕДОВАНИЕ, СЦЕПЛЕННОЕ С ПОЛОМ
(Х-ХРОМОСОМОЙ): ЦВЕТОВАЯ СЛЕПОТА
Наиболее часто встречающийся пример цветовой слепоты — неразличение красного и зеленого цветов (синдром, развивающийся в
результате недостатка соответ-
ствующего цвето-поглощаю-
щего пигмента в сетчатке глаза). Цветовая слепота встречается чаще у мужчин, чем у женщин. При изучении наследования цветовой слепоты были описаны два типа родословных: а) мать страдает цветовой слепотой, отец — нет, и все их сыновья (но ни одна из дочерей!) также имеют это
заболевание (рис. 3.1а), б) отец страдает цветовой слепотой,
мать и все дети имеют нормальное цветовое зрение, но один из внуков также цвето-слепой (рис. 3.16).
Феномен, объясняющий
тип наследования цветовой
слепоты, называется наследо-
79
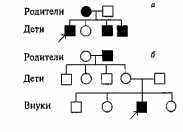
Рис. 3.1. Родословные пробандов, страдающих цветовой слепотой (признак наследуется по поло-сцепленному механизму).
В случае а основателем родословной, в которой цветовая слепота передается по наследству, является мать; в случае б — отец Обозначения те же, что и на рис. 2. 1
ванием, сцепленным с полом, — гены, ответственные за данную аномалию, локализованы в половых хромосомах. Поскольку особи разного пола несут разные половые хромосомы (XX у женщин и XY у мужчин), это приводит к определенным отклонениям от менделевских закономерностей наследования.
Цветовая слепота вызывается рецессивным аллелем с на Х-хро-мосоме. В результате того, что мужчины получают свою единственную Х-хромосому от матери, даже одного аллеля, вызывающего цве-тослепоту, достаточно, чтобы у мужчины, унаследовавшего аллель с на Х-хромосоме матери, развилось это заболевание. Для женщин же одной копии аллеля с недостаточно, они должны унаследовать две Х-хромосомы, несущие гены цветовой слепоты. Именно этим объясняется то, что у мужчин цветовая слепота встречается чаше, чем у женщин.
У человека существует пара хромосом, которая различается у мужчин и женщин. Женщины имеют две Х-хромосомы, а мужчины несут одну Х- и одну Y-хромосому. Y-хромосома значительно меньше по размеру, чем любая другая хромосома в геноме человека, и содержит «мужские гены», а также относительно небольшое количество генов, отвечающих за другие признаки. Сын и дочь наследуют одну хромосому X от матери; от отца дочери наследуют вторую Х-хромосому, а сыновья — Y-хромосому. Сыновья не могут унаследовать отцовскую Х-хромосому (если в зародыше сольются две Х-хромосомы — одна от матери, другая от отца, то это слияние и определит пол ребенка, т.е. разовьется женская особь). Дочери наследуют одну Х-хромосому от своих отцов, но для проявления рецессивных признаков они должны получить идентичную копию рецессивного аллеля от своих матерей.
Механизмы наследования цветовой слепоты показаны на рис. 3.2. Если семья состоит из цвето-слепой матери и нормального отца (рис. 3.2а), то это означает, что мать несет два аллеля с (по одному на каждой из Х-хромосом), а на Х-хромосоме отца располагается нормальный аллель С. Поэтому каждый из сыновей неизбежно унаследует одну из Х-хромосом матери, несущую с-аллель, и, соответственно, будет страдать цветовой слепотой. Все дочери тоже унаследуют одну из Х-хромосом матери, несущую аллель с, однако в результате того, что они получают Х-хромосому отца с нормальным аллелем С, фено-типически они будут нормальны, но будут носителями рецессивного признака (для обозначения фенотипически нормального носителя патологического аллеля символ этого индивидуума штрихуется наполовину). В случае, когда семья состоит из цвето-слепого отца и здоровой матери, не являющейся носителем рецессивного аллеля, фенотипически все дети здоровы (рис. 3.2б, первое поколение), но все дочери окажутся носителями аллеля цветовой слепоты, поскольку унаследовали отцовскую Х-хромосому, содержащую аллель с. Если же одна из дочерей образует семью с мужчиной, нормально различаю-