
Синтетические пептиды, взаимодействующие с различными типами холинорецепторов
| Вид материала | Автореферат |
- Пояснительная записка Цель полевой практики : знакомство с дикорастущими растениями, 212.83kb.
- Задача №9 «Жевательная резинка», 39.88kb.
- I межрегиональная выставка-ярмарка, 43.59kb.
- Самоопределение женщин с различными типами гендерной идентичности Иванова, 150.77kb.
- Лось различными исследователями, для провинций Северной Африки особенно характерно, 444.56kb.
- Искусство и религия, 72.78kb.
- Домашнее задание Требования к домашнему заданию: Необходимо создать несколько текстовых, 158.94kb.
- Синтетические пищевые красители, 68.81kb.
- Программа дисциплины Базы данных Семестры, 12.06kb.
- Алмазного Инструмента «томал», 592.31kb.
Синтез α-конотоксинов.
Синтез проводили как с использованием автоматического пептидного синтезатора Syro II для больших серий пептидов, содержащих замены различных остатков и варьирование длины первой цистеиновой петли, так и в ручном режиме для аналогов α-конотоксинов ImII, RgIA, PnIA, Vc1.1 и ArIB. Для активации защищенных аминокислот использовали 2 метода: DIPCDI/HOBt-метод для синтеза на автоматическом пептидном синтезаторе, и TBTU/DIPEA-метод для синтеза в ручном режиме. В реакциях конденсации использовали 3-5–кратные избытки активированной аминокислоты.
Для окисления линейных предшественников α-конотоксинов мы использовали подход как с одновременным, так и с поэтапным замыканием дисульфидных связей. В случае одновременного замыкания дисульфидных мостиков мы провели исследование влияния состава буферной смеси на выход целевого продукта. Для сравнения использовали смесь вода-ацетонитрил в соотношении 1:1 (для поддержания рН=8,5 в раствор добавляли диизопропилэтиламин) и буферную смесь, содержащую 10 mМ Tris-HCl и 0,2 mM EDTA (рН 8,5). В стандартную процедуру замыкания SS связей были внесены изменения: значение рН раствора пептида было увеличено с 7.5 до 8.5.
Увеличение рН позволило сократить время реакции с 36-72 до 18-35 ч, в случае применения для окисления дисульфидных связей водно-органической смеси в качестве основного продукта синтеза получали изомер с природным расположением дисульфидных связей. Процесс окисления контролировали при помощи теста Эллмана. Полученные α-конотоксины очистили с использованием препаративной ВЭЖХ. Все синтезированные соединения были охарактеризованы при помощи аналитической ВЭЖХ и масс-спектрометрии.
Для сравнения эффективности вышеуказанных методик окисления ряд пептидов был синтезирован с поэтапным замыканием дисульфидов. Было исследовано влияние порядка расположения ортогональных защитных групп остатков цистеина на выход целевого продукта, а также был применен метод удаления tBu-групп с помощью солей таллия (III) в трифторуксусной кислоте.
Индивидуальные особенности синтеза конкретных α-конотоксинов рассмотрены в следующих разделах.
Сравнение эффективности двух буферных смесей для замыкания дисульфидных связей в аналогах α-конотоксинов.
Окисление линейных предшественников α-конотоксинов [H5,R14]PnIA, [D7,L10]PnIA, [R5,L10,R14]PnIA, [Y5,R6,R7,L10,R14,W15]PnIA, [R5]Vc1.1, [GRCCS]MI(6-15), [R11]Vc1.1, [R5,D7]Vc1.1, [Y10]ImI, RgIA, ImII, [R5,D7]ImII, [L10 ]MI, [GCCS]MI(6-15), [GCCS, L10]MI(6-15) и [D7]Vc1.1 проводили в 50 мл буфера, содержащего 10 мМ Tris-HCl и 0,2 мМ EDTA при рН 8,5, при концентрации окисляемых пептидов 0,5 мг/мл. Для окисления пептидов [H5]PnIA, [L10,K14]PnIA, [D5,L10]PnIA, [R7, L10]PnIA, [R5,D7,L10]PnIA, [D5,R7,L10]PnIA, [D5,R7,V10]PnIA, [R5,D7,L10,R14]PnIA, MI и [D5,R7,L10,R14]PnIA использовали раствор ацетонитрила в воде в объемном соотношении (1:1) и концентрации пептида 0,5 мг/мл. Контроль за реакцией осуществляли при помощи обращенофазовой ВЭЖХ в градиенте ацетонитрила. Об окончании реакции судили по тесту Эллмана. Сравнение хроматографических профилей реакционных смесей показало большую эффективность метода с использованием смеси вода-ацетонитрил. Было отмечено, что скорость замыкания дисульфидных мостиков при использовании вышеуказанных буферных смесей практически одинакова (18-35ч.), однако выход целевого продукта был больше в случае использования водно-ацетонитрильной смеси. При окислении в водно-солевом буфере всегда наблюдалось образование нескольких дисульфидных изомеров и часто в сопоставимых количествах с целевым продуктом, в то время как при окислении в водно-органической фазе наблюдали образование одного изомера.
Исследование влияния расположения ортогональных защитных групп остатков цистеина на выход α-конотоксина RgIA.
В настоящее время отсутствуют литературные данные по исследованию оптимального расположения ортогональных защитных групп в синтезе α-конотоксинов. В большинстве работ в равной степени указываются оба способа расположения защит – tBu-группы (либо Acm и StBu) в положениях Cys1/Cys3, а также в положениях Cys2/Cys4. Синтез α-конотоксина RgIA был проведен на полимере Ринка с использованием Fmoc-схемы защитных групп аминокислот. Реакции конденсации проводили DIC/HOBt-методом с использованием 5-кратных избытков защищенной аминокислоты, при этом среднее время конденсации составляло 3 часа. В данной работе мы исследовали влияние порядка расположения ортогональных групп на выход α-конотоксина RgIA. Для этого мы синтезировали пептид RgIA, в котором тиольные функции остатков цистеина большой петли (Cys2/Cys4) имели тритильную защиту, а остатки Cys1/Cys3 защищены с помощью трет-бутильной группы, а также α-конотоксин RgIA, в котором остатки цистеина большой петли были защищены трет-бутильной группой, а малой - тритильной группой. Поскольку стратегия поэтапного замыкания дисульфидных связей подразумевает на первом этапе формирование SS-мостика между остатками цистеина содержащими свободные SH-группы, то в первом случае замыкание дисульфидной связи происходит с образованием петли, состоящей из 10 аминокислотных остатков, во втором случае петля состоит из 7 аминокислотных остатков (рис. 1):
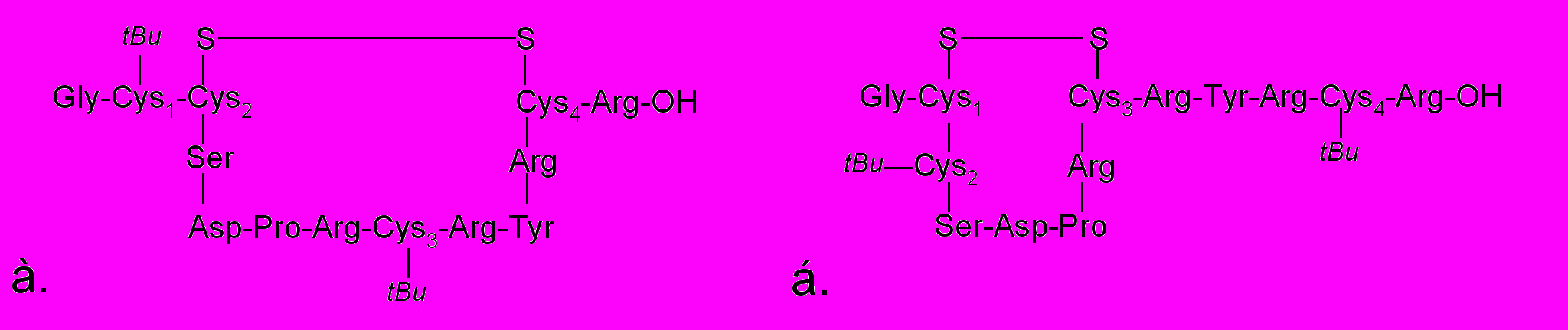
Рисунок 1. Строение 10- (а) и 7-членной петли (б) α-конотоксина RgIA.
Реакцию окисления проводили в 50% ацетонитриле, т.е. в условиях, оказавшихся оптимальными в случае синтеза с одновременным замыканием дисульфидов. Интересно, что формирование 10-членной петли проходило с выходом 75%, тогда как пептид, содержащий 7-членную петлю, получили с выходом 45%.
Для замыкания второй пары остатков цистеина использовали смесь дифенилсульфоксида с метилтрихлорсиланом в трифторуксусной кислоте в присутствии анизола в качестве скэвенджера. Поскольку α-конотоксины представляют собой бициклические соединения, для описания этой реакции, так же удобно использовать представление о петлях, содержащих разное количество аминокислотных остатков. Так при реакции образования второй дисульфидной связи (между остатками Cys1/Cys3) происходит формирование 7-членной петли, выход этой реакции составил 28%. Тогда как замыкание второй связи между остатками Cys2/Cys4 идет через формирование 10-членной петли с выходом 21%. Идентичность полученных пептидов доказали с помощью масс-спектрометрии, а также совместным элюированием.
По результатам данной работы установлено, что максимальный выход α-конотоксина RgIA достигается в случае расположения трет-бутильных групп в позициях Cys1/Cys3. При этом можно предположить, что на этапе формирования первой SS-связи высокий выход достигается благодаря меньшим конформационным напряжениям, которым подвержена 10-членная петля. Можно предположить, что более высокий выход реакции замыкания второго дисульфидного мостика между остатками Cys1/Cys3, обусловлен формированием жесткой циклической структуры, при этом остатки цистеина оказываются сближенными в пространстве, а остаток пролина в 6 положении, способствует закреплению конформации в положении близком к структуре пептида в окисленном состоянии.
Сравнение методов замыкания дисульфидных связей на примере синтеза двух изомеров α-конотоксина [Y10]ImII.
Синтез двух изомеров α-конотоксина [Y10]ImII (globular - дисульфидные связи между Cys2/Cys8 и Cys3/Cys12, ribbon –дисульфидные связи между Cys2/Cys12 и Cys3/Cys8) был осуществлен на полимере Ринка с использованием Fmoc-схемы защитных групп аминокислот. Тиольные функции остатков Cys2/Cys8 «природного» изомера и остатки Cys2/Cys12 «ribbon» изомера защищали с помощью tBu-групп. В качестве ортогональный защиты использовали кислотолабильные Trt-группы. Для активации защищенных аминокислот применяли TBTU/DIPEA-метод. Окисление первой дисульфидной связи линейных предшественников проводили в 50% ацетонитриле. Для замыкания второго SS-мостика использовали раствор трифторацетата таллия в трифторуксусной кислоте. Окисление проводили в трифторуксусной кислоте при 0ºС в течении 30 мин в присутствии 2 экв. анизола, который добавляли для связывания образующихся карбокатионов. После завершения реакции было обнаружено формирование сложного хроматографического профиля, однако MS-анализ реакционной смеси показал наличие одной молекулярной массы, соответствующей целевому продукту – α-конотоксину [Y10]ImII (рис. 2):
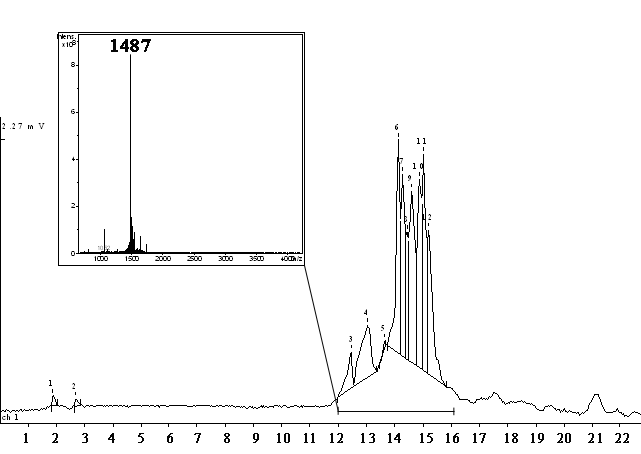
Рисунок 2. Хроматограмма α-конотоксина [Y10]ImII и масс-спектр смеси пептидных компонентов, образовавшихся после обработки раствором соли таллия (III).
Это обстоятельство позволило предположить, что в процессе деблокирования образовалась смесь комплексных соединений ионов таллия (I и III) c молекулами пептидов [Y10]ImII и [Y10]ImIIизо, которые разрушаются в процессе регистрации масс-спектра с образованием молекулярных ионов α-конотоксинов [Y10]ImII и [Y10]ImIIизо. Для проверки этого предположения мы провели качественную реакцию на ионы таллия с иодид ионом, который дает ярко-желтый осадок как с ионом Tl3+, так и Tl+ [17]. Однако добавление иодида калия не привело к видимым изменениям, что могло говорить, либо о высокой устойчивости комплексов, либо о неправомерности утверждения об их образовании. Поэтому мы провели предварительный кислотный гидролиз пептида соляной кислотой. После удаления избытка кислоты и добавления раствора иодида калия, наблюдали появление желтого осадка малорастворимых солей TlI3 и TlI. Чтобы исключить возможное окрашивание молекулярным иодом в смесь был добавлен бензол, который экстрагирует его из водного раствора. В качестве контроля мы использовали пептид [Y10]ImII, полученный по методике с одновременным замыканием дисульфидных связей, который после кислотного гидролиза не вызвал окрашивания при добавлении раствора иодида калия.
Случаи комплексообразования были описаны в литературе, однако, практических рекомендаций по разрушению подобных комплексов не приводится. Мы предложили использовать метод замещения лиганда (в данном случае пептида) на более сильный комплексообразователь ионов таллия. В качестве такого соединения выбрали этилендиаминтетрауксусную кислоту (ЭДТА), которая образует устойчивые хелатные комплексы с ионами тяжелых металлов, выступая в качестве пента- или гексадентатного лиганда. Для разрушения комплексных соединений к пептидам, содержащим одну дисульфидную связь, добавили 1 М раствор ЭДТА. Для исключения возможности дисульфидного обмена, использовали раствор с рН 4,5. Как показано на рис. 3, с течением времени наблюдалось постепенное уменьшение числа хроматографических пиков, вплоть до образования одного вещества (через 56ч.) с молекулярной массой 1485.7 Da (расчетная молекулярная масса α-конотоксина [Y10]ImII 1485.6 Da) время удерживания которого в дальнейшем не изменялось.
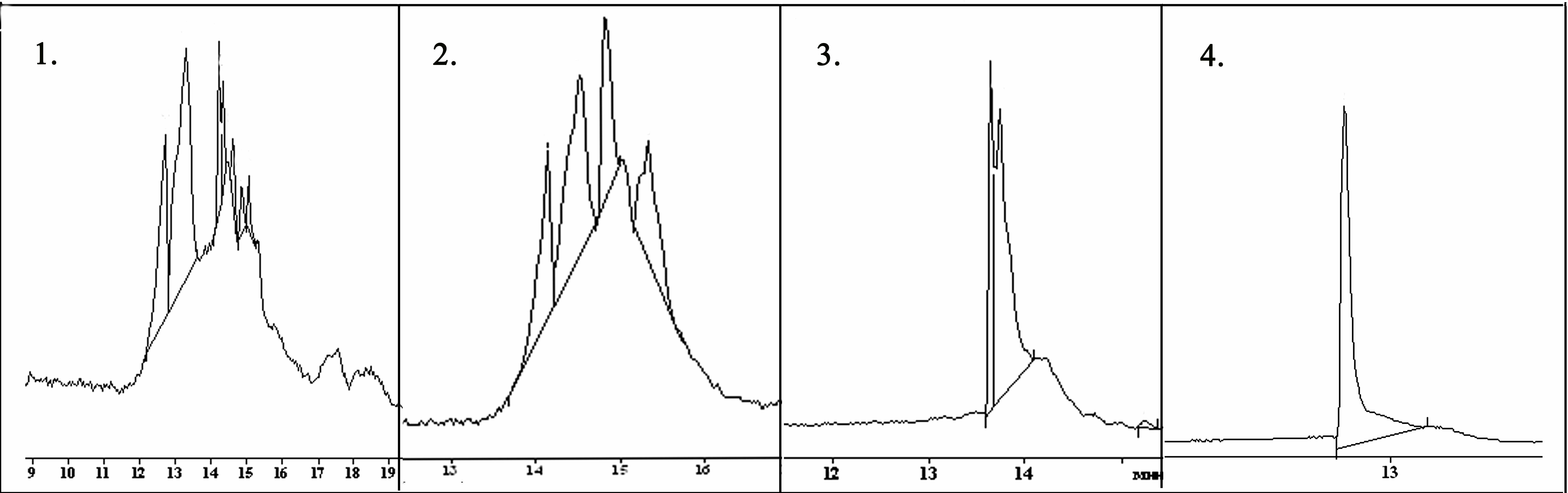
Рисунок 3. Разрушение комплексных соединений ионов таллия с пептидом [Y10]ImII. Через 5 мин. после добавления ЭДТА ( 1), через 16 часов (2), через 32 часа (3) и через 56 часов (4).
Следует отметить, что выходы пептидов на этапе окисления трифторацетатом таллия были достаточно высокими – 73% для globular-изомера, и 60% в случае ribbon-изомера. Это примерно в 2-3 раза превышает выходы целевого продукта по сравнению с использованием смеси дифенилсульфоксида с метилтрихлорсиланом для окисления второй дисульфидной связи α-конотоксинов RgIA, Vc1.1, [Y0]MII, [L11, D16 ]ArIB, [S0, L11, D16]ArIB и [(SGGG)0, L11, D16]ArIB (синтез этих пептидов рассмотрен в следующих разделах). Выходы globular-изомера α-конотоксина [Y10]ImII составил 15% в расчете на стартовую аминокислоту, ribbon-изомера 8%.
Для наработки препаративных количеств α-конотоксина [Y10]ImII мы провели синтез с использованием одновременного замыкания дисульфидных связей. Окисление проводили в 50% ацетонитриле, что привело к образованию одного основного продукта. В результате совместного элюирования в условиях аналитической ВЭЖХ продуктов реакции одновременного замыкания дисульфидных связей с globular-изомером, который был получен с использованием поэтапного замыкания дисульфидов был идентифицирован изомер с природным расположением дисульфидных мостиков. Выход α-конотоксина [Y10]ImII, синтезированного с использованием одновременного замыкания дисульфидных связей, составил 33%.
Синтез α-конотоксинов Vc1.1, [Y0]MII, [L11, D16 ]ArIB, [S0, L11, D16]ArIB и [(SGGG)0, L11, D16]ArIB.
Синтезы α-конотоксинов Vc1.1 и [Y0]MII были осуществлены на полимере Ринка, а [L11, D16 ]ArIB, [S0, L11, D16]ArIB и [(SGGG)0, L11, D16]ArIB на полимере Ванга. Для защиты функциональных групп использовали Fmoc-схему. Синтез проводили с использованием поэтапного замыкания дисульфидных связей. На основании экспериментов, описанных в предыдущем разделе, выбрали оптимальный порядок расположения защитных групп остатков цистеина - Cys1/Cys3 содержали tBu-группы, а Cys2/Cys4 - тритильные группы. Деблокирование с одновременным отщеплением пептида от полимера проводили в одну стадию с использованием смеси трифторуксусной кислоты с анизолом, водой и этандитиолом в качестве скевенджеров. По данным аналитической обращено-фазовой ВЭЖХ содержание линейных предшественников α-конотоксинов после реакции деблокирования во всех синтезах составило более 70%
Для замыкания первой дисульфидной связи, между остатками Cys2/Cys4, использовали метод окисления кислородом воздуха в 50% растворе ацетонитрила. Для замыкания второй дисульфидной связи использовали смесь дифенилсулфоксида с метилтрихлорсиланом в трифторуксусной кислоте, содержащей анизол в качестве ловушки карбокатионов.
В заключение этого раздела необходимо отметить, что предлагаемая нами схема синтеза α-конотоксинов разрабатывалась в течение нескольких лет на примере синтеза пептидов, необходимых для наших исследований ацетилхолиновых рецепторов, и неоднократно видоизменялась, вбирая в себя черты как работ зарубежных авторов, так и наше видение оптимальных путей синтеза. В конечном итоге, основные черты нашего подхода к синтезу природных α-конотоксинов (но не их аналогов) заключаются в следующем:
1. Для защиты тиольных функций всех остатков цистеина использовать кислотолабильные защитные группы.
2. В случае получения линейного продукта высокой степени чистоты целесообразно не применять предварительную очистку, а сразу проводить реакцию замыкания дисульфидных связей.
3. Оптимальным для замыкания дисульфидных мостиков является использование смеси вода-ацетонитрил в объемном соотношении 1:1, при рН 8,5-9. Это позволяет уменьшить время окисления и получить целевой продукт с высоким выходом.
Можно полагать, что эта схема окажется полезной и для получения других биологически активных коротких пептидов, содержащих две дисульфидные связи – таких, как апамины или эндотелины. Эта схема в целом пригодна и для синтеза аналогов α-конотоксинов, если в распоряжении исследователей имеются возможности с помощью структурного анализа или функциональных тестов идентифицировать изомер с природным расположением дисульфидных связей. Однако при отсутствии такой возможности оправдано использование ортогональных защит тиольной группы остатков цистеина и последовательное замыкание дисульфидных связей. Большим недостатком подхода, использующего ортогональные защиты, является низкий выход целевого продукта. Вследствие этого он мало подходит для синтеза препаративных количеств пептида, необходимых для проведения углубленных структурно-функциональных исследований. Поэтому нам кажется оптимальным использовать комбинацию этих подходов: для этого препаративные количества пептида следует получать с использованием одновременного замыкания дисульфидных связей, а для установления порядка расположения дисульфидных связей в полученных изомерах использовать совместное элюирование с пептидом, полученным по методу поэтапного замыкания SS-мостиков в аналитических количествах.
Исследование влияния структуры на биологическую активность α-конотоксинов и их аналогов.
Определение биологической активности аналогов α-конотоксина MI.
α-Конотоксин MI принадлежит к классу токсинов, связывающихся с мышечными холинорецепторами. Общим для этого класса является наличие 3 аминокислотных остатков в первой цистеиновой петле. С другой стороны, α-конотоксины, действующие на нейрональные нАХР (например ImII, MII, PnIA, GID) имеют по 4 остатка в первой петле. Можно предположить, что увеличение длины первой цистеиновой петли «мышечных» α-конотоксинов может привести к увеличению сродства полученных аналогов к нейрональным подтипам нАХР. Для удлинения первой петли мы ввели остаток серина в положение 5 токсина MI.
Биологическую активность α-конотоксина MI и его аналогов определяли по их способности взаимодействовать с мембраносвязанным никотиновым ацетилхолиновым рецептором из электрического органа ската Torpedo сalifornica. Для определения того, насколько увеличение длины первой цистеиновой петли повлияет на селективность пептида MI, была исследована способность взаимодействовать с нейрональным рецептором - α7 подтипом нАХР, трансфецированным в клетки GH4C1 (табл. 2).
Для проведения анализов без введения дополнительных модификаций в исследуемые соединения использовали метод конкурентного радиолигандного анализа, который основан на способности исследуемых аналогов подавлять связывание иодированного производного α-бунгаротоксна ([125I]Bgt) с препаратами АХР2. Для этой цели было синтезировано радиоактивное производное α-бунгаротоксина с введенным в остаток Tyr радиоактивным изотопом иода. В работе было использовано только моно-[125I]-иодированное производное α-бунгаротоксина.
Ингибирующая активность всех полученных аналогов α-конотоксина MI в концентрации ≈ 0,003 мМ представлена в Табл. 2.
Таблица 2. Ингибирующая активность аналогов α-конотоксина MI.
| α-конотоксин | Ингибирующая активность ( Torpedo сalifornica), % | Ингибирующая активность (α7), % |
| MI | 89 | 1 |
| [L10]MI | 62 | 5 |
| [GRCCS]MI(6-15) | 73 | 2 |
| [GCCS]MI(6-15) | 34 | 1 |
| [GCCS, L10]MI(6-15) | 20 | 1 |
Из приведенных данных видно, что наибольшую ингибирующую активность на рецепторе из Torpedo сalifornica проявил α-конотоксин МI. Кроме того, достаточно высокую активность показал аналог с увеличенной длиной первой цистеиновой петли [GRCCS]MI(6-15). Следует отметить, что большое значение для активности имеет остаток аргинина во 2 положении, поскольку его удаление приводит к резкому падению активности пептидов [GCCS]MI(6-15) и [GCCS, L10]MI(6-15).
В тоже время α-конотоксин [GRCCS]MI(6-15), имеющий увеличенную длину первой петли, а также аналоги [GCCS]MI(6-15) и [GCCS, L10]MI(6-15) не проявили высокого сродства к нейрональному подтипу холинорецептора.
Замена остатка лизина в 10 положении α-конотоксина MI на лейцин несколько ухудшила сродство полученного аналога к холинорецептору Torpedo сalifornica по сравнению с немодифицированным пептидом. Однако, сродство этого аналога ([L10]MI) к нейрональному подтипу холинорецептора было наибольшим среди синтезированных аналогов, хотя его активность была значительно ниже, чем у «нейрональных» α-конотоксинов.
Таким образом было показано, что увеличение длины первой цистеиновой петли не приводит к увеличению сродства соответствующих аналогов α-конотоксина MI к α7 подтипу нАХР.
Определение биологической активности аналогов α-конотоксина PnIA.
Для изучения влияния заряженных и гидрофобных аминокислотных остатков на сродство аналогов α-конотоксина PnIA к α7 полтипу нАХР, был синтезирован ряд аналогов, в которых остаток в определенном положении пептидной цепи, несущий заряд, заменяли на гидрофобный остаток, либо заменяли на противоположно заряженный остаток.
Для исследования изменения сродства к α7 нАХР мы синтезировали ряд пептидов, в которых в качестве основы была выбрана последовательность пептида [A10L]PnIA.
2 Эта часть работы была выполнена совместно с д.х.н. И.Е. Кашеверовым в лаборатории лиганд-рецепторных взаимодействий
Также мы синтезировали пептиды, не несущие этой базовой мутации [A10L], но имеющие положительный заряд в 5 и 14 положениях - [H5]PnIA и [H5,R14]PnIA. Исследование их биологической активности проводили на α7 подтипе нАХР, трансфецированном в клетки GH4C1, а также на ацетилхолин-связывающих белках из A. californica и L. stagnalis.
Ингибирующая активность всех полученных аналогов α-конотоксина PnIA в концентрации ≈ 0,003 мМ представлена в Табл. 3: