
Высоких Технологий "ХимРар"
| Вид материала | Реферат |
- Инженерно- физический факультет высоких технологий Инженерно-физический факультет высоких, 561.11kb.
- Отчет о выполнении государственного задания за 2010 год отчет ау «Технопарк высоких, 1094.59kb.
- Положение о присвоении статуса технопарка в рамках Некоммерческого партнерства «Ассоциация, 88.27kb.
- 11-я Международная выставка-ярмарка высоких технологий «chtf-2009», 49.38kb.
- Xi международная научно-практическая конференция "Фундаментальные и прикладные исследования, разработка, 39.29kb.
- Всовременном мире благосостояние населения и могущество государства зависят от науки,, 190.35kb.
- Венчурные фонды в экономике высоких технологий, 114.8kb.
- Ровождающейся интенсивными темпами внедрения высоких технологий, одной из основных, 71.9kb.
- Семинар ярмарка "российские технологии для индустрии", 30.09kb.
- Инновационная активность в сфере высоких технологий для расширения перспективы предприятия, 63.93kb.
2. Экспериментальная оценка основных физико-химических показателей
Органолептическое испытание. Внешний вид, цвет и запах.
Белок «Йодказеин» представляет собой неоднородный порошок рыжего цвета с коричневыми включениями разнородных по размеру гранул. Порошок обладает резким запахом йода и органических растворителей.
Белок «Биойод» имеет вытянутую кристаллическую структуру порошка после лиофильной сушки с кремовым светло-желтым оттенком и запахом молока.

Рис. 3. Внешний вид йодированных молочных белков «Йодказеин» (слева) и «Биойод» (справа)
Сульфатная зола.
Сульфатная зола из 1,0 г (точная навеска) субстанции является важным критерием, указывая на неорганический остаток после озоления (ГФ XII, ч.1, с.115).
1,0 г субстанции (точная навеска) помещают в предварительно прокаленный и точно взвешенный кварцевый тигель, добавляют 1,0 мл серной кислоты концентрированной и осторожно нагревают на электрической плитке для удаления серной кислоты. Температуру нагрева на электрической плите последовательно повышают от 180 °С до 300 °С с шагом 50 °С во избежание обильного вспенивания и потери вещества из аналитического объема. Затем тигель помещают в муфельную печь при температуре 300 °С и медленно нагревают до 500 °С. Тигель с веществом прокаливают до постоянной массы при 500 °С. По окончании прокаливания, тигель охлаждают в эксикаторе. К остатку в тигле добавляют 1,0 мл серной кислоты концентрированной, повторяют процедуру с прокаливанием, по окончании взвешивают остаточную массу.
Вода по методу К. Фишера (волюмометрический метод)
Содержание воды в субстанции служит важным критерием о гидрофильных свойствах йодированного белка, косвенно характеризуя состояние белка после обработки в процессе получения.
Титратор: Mettler Toledo V30
Титрант: КФИ-1К Т5
Растворитель: КФ-РТ-Кетон
Титр: T= 5.04414 мг/мл
Методика определения:
Объёмометрическое титрование воды, содержащейся в пробе, проводили по методу К. Фишера (полумикрометод) с потенциометрической индикацией конечной точки титрования на поляризованном платиновом электроде (ГФ XI, вып. I, с. 176).
Около 0,1 г (точная навеска) субстанции титруют реактивом К.Фишера (с предварительно установленным титром).
Растворитель для титрования используют безметанольный во избежание побочных реакций среды растворения с белковой матрицей.
Перед началом титрования проба перемешивается и растворяется в течение 45 с.
Расчет проводят по формуле:
Rx=(Vэкв.*T-t*D/1000)*C/m,
где Vэкв. – объём титранта, пошедший на титрование воды в навеске, мл;
m – масса навески, г;
T – концентрация титранта КФИ-1К T 5, г/мл (титр).
t – время титрования, мин
D – относительный дрейф, мкг/мин
C =0.1 - поправочный фактор
Результаты содержания воды приведены в сравнительной таблице йодированных белков.
3. Содержание молекулярного йода, неорганического йодид-иона и ковалентно связанного органического йода
3.1 Методика определения свободного молекулярного йода. Методические указания МУК 4.1.1090—02
Определение молекулярной формы йода в йодированных белках выполнено в соответствии с ГОСТ Р 53751-2009 на молочные продукты и продукты детского питания на молочной основе.
Определение проводили титриметрическим методом с предварительной пробоподготовкой на основе экстракции и выделении свободного йода в спиртовую фракцию, далее оттитровываемого натрием серноватистокислым, по расходу которого расчетным путем определяют содержания свободного йода в пробе.
Перед выполнением измерений проводят следующие работы: приготовление растворов, отбор проб.
Все растворы готовятся на безйодной дистиллированной воде. Дистиллированная вода перегоняется в присутствии К2СО3. Спирт ректификат перегоняется в присутствии К2СО3.
Существующая ранее фотометрическая методика определения ( Сборник методических указаний. МУК 4.1.737—99—4.1.754—99) из-за недостаточной чувствительности не позволяет контролировать содержание йода в воде на уровне предельно допустимой концентрации (ПДК 0,125 мг/дм3). Существенным недостатком йодометрической методики является отсутствие метрологической аттестации.
Методические указания, в соответствие с которыми анализировались пробы йодированных белков, дают возможность устанавливать содержание молекулярного йода в диапазоне концентраций 0,01—1 мг/дм3. Метод метрологически аттестован и обеспечивает определение молекулярного йода с пределом обнаружения 0,08 ПДК.
Методика обеспечивает выполнение измерений с погрешностью, не превышающей ± 30 %, при доверительной вероятности 0,95.
Данная методика измерения концентрации йода титриметрическим методом разработана для определения йодидов в водах и основана на окислении йодидов до йодатов в кислой среде бромной водой с восстановлением последних до свободного йода по формуле:
Г +3 Вг2 + ЗН2О = IO-3 +6Н+ +6Вг-; (1)
КIO3 + 5 KI + 3H2S04 = ЗI2+ 3K2S04+ ЗH2О; (2)
I2 + 2Na2S203 = Na2S406+ 2NaI. (3)
В исследовании свободного молекулярного йода титриметрическим методом использовали количественное титрование тиосульфатом натрия, взаимодействие протекает уравнению (3).
Количественное определение проводят йодометрическим титрованием. Нижний предел измерения йода в анализируемой пробе 10 мкг. Определению не мешают другие галогены.
Подготовка стеклянной посуды
Новую стеклянную химическую посуду первоначально промывают смесью концентрированной серной кислоты и перекиси водорода (в соотношении примерно 10:1 по объему), затем – раствором концентрированной азотной кислоты, разбавленной бидистиллированной водой (в соотношении примерно 1:10 по объему). После обработки растворами кислот посуду промывают бидистиллированной водой несколько раз.
При последующем использовании стеклянную посуду промывают раствором концентрированной азотной кислоты, разбавленной бидистиллированной водой (в соотношении примерно 1:10 по объему), затем бидистиллированной водой.
Новые кварцевые стаканчики или фарфоровые чашки обрабатывают раствором концентрированной азотной кислоты, разбавленной бидистиллированной водой (в соотношении примерно 1:10 по объему), затем прокаливают в муфельной печи при температуре (550 + 50) °С в течение (30 + 1) мин.
Пробы с низким содержанием йода (менее 0,01 мг/л) предварительно концентрируют упариванием. Для определения отбирают такой объем пробы, чтобы содержание в нем йода было в пределах 0,01—1 мг. В термостойкий стакан помещают пробу исследуемого белка массой около 1,00 г (точная навеска), добавляют достаточное количество бидистиллированной воды для растворения, прибавляют 10 капель 1 %-ного раствора фенолфталеина и раствор К2СО3 до ярко красного окрашивания, не исчезающего при помешивании.
В делительную воронку вместимостью 50 см3 помещают раствор испытуемого йодированного белка, добавляют 20 см3 бидистиллированной воды и 20 см3 этилового спирта, встряхивают 30 мин на лабораторном перемешивающем устройстве. Экстракцию спиртом проводят до тех пор, пока слой растворителя не станет бесцветным. Верхнюю водную фракцию переносят в фарфоровую чашку и высушивают в сушильном шкафу при температуре 150 °С для дальнейших испытаний на содержание связанного йода.
Полученный экстракт выпаривают на водяной бане, прибавив 2 капли концентрированного раствора К2СО3. После этого чашку просушивают в сушильном шкафу и прокаливают в электропечи. Так как в экстракте минеральных веществ мало, в этих условиях происходит быстрое и полное сгорание всего органического вещества. После охлаждения чашки добавляют 3—4 капли дистиллированной воды и опять экстрагируют небольшими порциями спирта (10 см3). Экстракт осторожно концентрируют на водяной, не сильно нагретой бане с таким расчетом, чтобы спирт в чашке не закипел.
Добавляют кристаллик химически чистого йодистого калия, полученного и подкисляют двумя каплями серной кислоты. Прибавляют три капли 0,5 %-ного раствора крахмала, свежеприготовленного, и титруют 0,001 моль/дм3 раствором серноватистокислого натрия, приготовленного до изменения окраски раствора, не исчезающей в течение 1 мин.
Контрольная пробу, готовят, используя вместо пробы йодированного белка 20 см3 бидистиллированной воды. Далее проводят титрование как описано выше.
Обработка результатов измерений
Содержание йода Х, мкг/кг, вычисляют по формуле
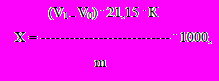 , (1)
, (1)где:
V1 – объем раствора серноватистокислого натрия молярной концентрацией
0,001 моль/дм3, израсходованного на титрование в исследуемой пробе, см3;
V0 – объем раствора серноватистокислого натрия молярной концентрацией
0,001 моль/дм3, израсходованного на титрование в контрольной пробе, см3;
21,15 – масса йода, соответствующая 1 см3 раствора серноватистокислого
натрия молярной концентрацией 0,001 моль/дм3, мкг;
К – коэффициент поправки к титру 0,001 моль/дм3 раствора
серноватистокислого натрия;
m – навеска исследуемой пробы , г;
1000 – коэффициент пересчета результатов на 1000 г продукта;
За окончательный результат принимают среднеарифметическое значение результатов двух параллельных измерений, округленное до первого десятичного знака, если выполняется условие приемлемости.
Приписанные характеристики погрешности и ее составляющих метода определения содержания йода при Р = 0,95 приведены в таблице ниже:

Результаты испытания йодированных белков на содержание молекулярного йода.
Результаты измерения проб йодированных белков «Йодказеин» и «Биойод», проведённые по методике описанной выше выявили наличие 0,42 масс.% свободного йода в пробе белка «Йодказеин».
В пробе йодированного белка «Биойод» йод в свободной молекулярной форме не обнаружен.
Методика выполнения измерений (МУ 31-07/04) содержания йода в пищевых продуктах, продовольственном сырье, кормах и продуктах их переработки, лекарственных препаратах, витаминах, БАДах, биологических объектах (моча) методом инверсионной вольтамперометрии.
Методика выполнения измерений разработана в лаборатории приборов вольтамперометрического анализа химико-технологического факультета Томского политехнического университета и ООО «НПП Томьаналит» и аттестована в соответствии с ГОСТ Р 8.563. Методика определения согласовывается и не противоречит описанию методике вольтамперометрического детектирования йодид-ионов ГОСТ Р 53751-2009 на молочные продукты и продукты детского питания на молочной основе, а также вольтамперометрическому определению йода в пищевых продуктах МУК 4.1.1187-03.
Определение иодид-ионов методом инверсионной вольтамперометрии проводили на ртутно-плёночных и серебряных электродах. Сущность метода заключается в предварительном электроконцентрировании иодид-ионов на поверхности электрода при постоянном потенциале в виде малорастворимого соединения (иодида ртути или иодида серебра) и последующем электрорастворении осадка с поверхности электрода.
Обеспечить удовлетворительную воспроизводимость результатов на серебряных электродах удается только при сложной регенерации поверхности, которая обычно включает в себя стадии механической, химической и электрохимической обработки. В данном исследовании в качестве индикаторного электрода использовали ртутно-пленочный электрод (РПЭ).
Методика включает в себя предварительную подготовку проб путем минерализации и последующий анализ водного раствора минерализованной пробы методом катодной инверсионной вольтамперометрии.
Пробоподготовка
Разработаны и апробированы две методики пробоподготовки йодированных белков для последующего вольтамперометрического определения йодид-ионов:
- микроволновой минерализации действием смеси концентрированных HNO3 и H2O2 в кварцевом реакторе закрытого типа на микроволновой системе CEM Discovery
- «мокрого» щелочного озоления с добавками навески пробы йодированного белка на нагревателях (электрическая плита, муфельная печь) открытого типа.
Предварительная пробоподготовка в первом случае заключалается в растворении пробы йодированного белка в воде в соответствии с техническим условиями на каждый йодированный белок при незначительном нагреве до 60 ˚С и перемешивании.
Далее раствор белка переносят в герметичный кварцевый реактор для термообработки микроволновым способом. В сосуд для минерализации объёмом 10 мл. вносили 3 мл. концентрированной HNO3 и 1 мл. концентрированной H2O2. Реактор герметично закрывался крышкой и помещался в зону для микроволновой обработки.
Температура и давление в реакционной зоне контролировались аппаратно (рис.1) в интервале температур 160-180 ˚С и давления 10-17 атм.
Время обработки рекомендовано в интервале 30-35 мин и зависит от матричных эффектов связывания. После реактор охлаждали, раствор после минерализации испытывали методом постояннотоковой вольтамперометрии на общее валовое содержание йодид-ионов в соответствии с методическим указанием МУ 31-07/04.

Рис.4. Микроволновая минерализация проб йодированных белков в окислительной смеси HNO3-H2O2.
Подготовка проб в случае «мокрого» щелочного озоления включает следующие операции: первичную оценку содержания несвязанного йодид-иона, окклюдированного в белковой матрице и суммарное валовое содержание йода после муфельного щелочного озоления этой же пробы.
Для этого перед анализом пробу образца навеской 0,1-0,2 г., взвешенной на аналитических весах с точностью 0,0002 г, помещают в кварцевый стаканчик (фарфоровый тигель), вместимостью 25 см3, добавляют (4-5) см3 раствора калия гидроокиси концентрации 10 % массовых и оставляют на 30 минут. Добавляют 15 см3 бидистиллированной воды, тщательно перемешивают раствор до полного растворения образца. Для анализа берут аликвоту пробы не более 0,01 мл.
Далее в кварцевый стаканчик после предварительной подготовки и первичной оценки несвязанного йода добавляют 1 см3 раствора калия гидроокиси концентрации 10 % массовых и выпаривают на электроплитке при температуре 120 ˚С в течение 40-60 минут, накрыв стаканчик фильтровальной бумагой. Затем выдерживают стаканчики в муфеле или в камере озоления печи 30–40 минут при 550 ˚С.
Полного разрушения органических веществ можно достигнуть таким озолением пробы, но при этом возможно улетучивание йода. Для предотвращения потерь предварительно пробу выдерживали с 10 % раствором гидроксида калия в течение более длительного времени (1-2 ч.). После этого раствор также выпаривают до сухих солей и выдерживают в муфельной печи при температуре 480 ˚С 15 минут. Для ускорения процесса разложения к пробе добавляют раствор нитрата калия 1 моль/дм3, выпаривают и снова выдерживают в муфельной печи при той же температуре. Недостатком этого способа является длительность – до 3 часов, а также возможность загрязнения пробы используемыми реактивами.
Данный подход двухкратной обработки одной навески пробы позволяет двумя независимыми способами минерализации - микроволновым и муфельным - установить методом катодной ИВА содержание несвязанного свободного и органически связанного ковалентного йода в аналитически определяемой форме йодид-ионов.
Таким образом, в процессе минерализации пробы и последующем ультрафиолетовом облучении раствора минерализованной пробы переводят все формы йода в иодид-ионы.
Эффективность применения каждого из методов пробоподготовки (микроволнового и муфельного) оценивали по возможности получения воспроизводимого аналитического сигнала, обеспечивающего количественную оценку содержания общего йода в пробе. Правильность оценки проверяли методом «введено-найдено» при введении в анализируемый раствор аттестованной смеси ГСО.
Методика измерения
Иодид-ионы концентрируют на серебряном модифицированном или ртутно-плёночном электродах в виде малорастворимого осадка с последующим катодным восстановлением осадка при линейном изменении потенциала. Возникающий при этом катодный пик при потенциале минус (0,4±0,05) В для серебряного электрода модифицированного и минус (0,3±0,05) В для ртутно-плёночного электрода является аналитическим сигналом. Содержание иодид-ионов в растворе подготовленной пробы определяется по методу добавок аттестованной смеси иодид-ионов.
Измерения проводили на компьютеризированном вольтамперометрическом анализаторе ТА-4 производства ООО НПП «Томьаналит» (г.Томск) с встроенной УФ-лампой (λ=185-260 нм; Р=20 Вт) и трехэлектродной электрохимической ячейкой: вспомогательный электрод и электрод сравнения - хлоридсеребряный электрод (в 1 М KCl). Индикаторный РПЭ готовили «механическим» нанесением плёнки ртути или путем электролитического осаждения ртути в режиме заданного тока на серебряную проволоку диаметром 1,1 мм и длиной 8-9 мм, запрессованную в полимерный стержень, из насыщенного раствора Hg2(NO3)2 при постоянном токе 1,5 мА в течение 600 с. Перемешивание анализируемых растворов осуществляли путем вибрации индикаторного электрода. Все используемые реактивы были марки о.с.ч. и не подвергались дополнительной очистке. Все растворы готовили на бидистиллированной воде.
Растворы иодид-ионов готовили разбавлением государственных стандартных образцов ГСО.
Электроосаждение иодидов на поверхность РПЭ проводили в виде малорастворимого соединения со ртутью Hg2I2 при постоянном потенциале, при котором происходит электрорастворение металлической ртути. Растворение осадка проводили при постояннотоковой или переменнотоковой форме развертки поляризующего напряжения от минус 0,05 В до минус 0,95 В со скоростью 100 мВ/с. При этом на вольтамперограмме регистрировали катодный пик при потенциале минус (0,35±0,05) В, который служил аналитическим сигналом иодид-ионов. Для сравнения другие формы йода, например йодат-ион восстанавливается при потенциале минус (1,4±0,05) и возможно раздельное их обнаружение в совместном присутствии (рис.5).

Рис .5 Типичный вид вольтамперограмм форм йода в виде йодат и йодид анионов, позволяющая их раздельное обнаружение.
Применение дифференциально-импульсной развертки потенциала (амплитуда импульса - 30 мВ, ширина импульса – 25 мс) позволяет увеличить величину аналитического сигнала в 2,5-3 раза. Содержание иодид-ионов в пробе определяли по методу добавок. Мешающее влияние растворенного кислорода устраняли путем барботажа азота через анализируемый раствор или фотохимическим способом.
В качестве фоновых электролитов использовали муравьиную кислоту. Величина аналитического сигнала иодид-ионов не зависит от концентрации муравьиной кислоты в диапазоне от 0,05 до 0,8 моль/дм3.
Использование в качестве фонового электролита муравьиной кислоты позволяет избежать применения инертного газа и проводить дезактивацию растворенного кислорода за счет протекания фотохимической реакции при УФ-облучении анализируемого раствора.
Таблица - Параметры подготовительных этапов (стадий) методики «Определение йодав продуктах»
| Этап | Потенциал, В | Время выполнения этапа, с | Состояние исполнительных устройств | |
| УФ-лампа | Вибрация | |||
| Подготовка* | -0.5 | 60 | Вкл. | Вкл. |
| Растворение | -0.5 | 10 | Вкл. | Вкл. |
| Накопление | -0.1 | 30 | Вкл. | Вкл. |
| Успокоение | 0 | 2 | Выкл. | Выкл. |
* Этап «Подготовка» выполняется только перед регистрацией серии вольтамперограмм.
Потенциал пика йода: минус 0.4 В для СЭМ; минус 0.3 В для РПЭ.
Метод измерения: постояннотоковый.
Потенциал начала развертки: 0 В.
Потенциал конца развертки: минус 1.0 В.
Скорость развертки: 100 мВ/с.
Метод расчета пиков: по высоте.
Инверсия кривых: включена.
Параметры отмывки: по методике, пропуская этап «Накопление», число повторов – 5.
Параметры подготовки (нанесение пленки ртути на РПЭ): канал «А»; ток 1,5 мА; время 180 секунд; вибрация выключена (установлено значение «0»).
Проверка работы электродов осуществляется методом «введено-найдено»
Проверку работу электродов проводят:
а) ежедневно перед началом работы;
б) после нанесения пленки ртути на поверхность электрода;
в) при расхождении результатов параллельных определений свыше допускаемого;
г) при отсутствии на вольтамперограммах пика йода.
В кварцевые стаканчики с фоновым раствором, проверенные на чистоту добавляют 0,04 см3 аттестованной смеси иодид-иона концентрации 10 мг/дм3.
Устанавливают время на этапе «Подготовка» 20 с.
Облучение раствора в течение 60 секунд для дезактивации растворенного кислорода проводят только один раз. При последующих регистрациях в этом же растворе, если после облучения раствора прошло не более пяти минут, и если в облучаемый раствор не вносятся компоненты, мешающие определению, уменьшают время подготовки до 20 секунд.
Устанавливают параметры пробы:
Объем аликвоты минимальный для обнаружения сигнала: 0,005 см3.
Объем минерализата: в зависимости от выбранного способа минерализации составляет от 5,0 см3 для микроволновой минерализации до 10 см3 для щелочного озоления.
Масса навески по каналам: 0,05-0,2 г.
Размерность для расчета концентраций - мг/кг.
Проводят регистрацию вольтамперограмм пробы в масштабе 10:1 – 20:1.
После регистрации исключают, если необходимо, невоспроизводимые вольтамперограммы. Количество воспроизводимых вольтамперограмм в каждом окне должно быть не менее двух. В противном случае регистрацию повторяют.
Обрабатывают полученные вольтамперограммы пробы: усредняют и при необходимости корректируют разметку линии остаточного тока.
Устанавливают параметры добавки аттестованной смеси: концентрация – 10 мг/дм3; объем - 0,04 см3.
Вносят в каждую ячейку 0,04 см3 аттестованной смеси иодид-иона концентрации 10 мг/дм3 и запускают регистрацию вольтамперограмм пробы с добавкой.
Получают две-три воспроизводимые вольтамперограммы пробы с добавкой. После регистрации исключают, если необходимо, невоспроизводимые вольтамперограммы. Количество воспроизводимых вольтамперограмм в каждом окне должно быть не менее двух. В противном случае регистрацию повторяют. Обрабатывают полученные вольтамперограммы пробы с добавкой: усредняют и при необходимости корректируют разметку линии остаточного тока.
Выполняют команду «Расчет». При наличии на вольтамперограммах фона пика йода, включают «Учет фона». Если полученные значения концентрации йода входят в интервал 0,030-0,050 мг/дм3, то электроды работают удовлетворительно. Если расхождение между полученной концентрацией и введенной превышает 25 % (например, 0,025 мг/дм3 - полученная, 0,040 мг/дм3 - введенная), проверку электродов повторяют с новым фоновым раствором.
Содержание йод-органических соединений определяют по разнице валового содержания йода и суммарного содержания иодид- и иодат-ионов. Для определения валового содержания йода использовали описанные методы пробоподготовки: микроволновую минерализацию и щелочное озоление с добавками.
Адекватность и достоверность определения йода в йодированных белках проверяли по содержанию йода в свежей йодированной соли с заявленным содержанием 40±15 мг/кг.
В приложении 1 приведена типичная серия вольтамперограмм катодной ИВА для раствора соли «Зимушка краса» (Нидерланды).
Результаты содержания органической связанной формы йода и свободной неорганической для исследуемых йодированных белков приведены в сравнительной таблице (с.40-41).
Прямой метод определения валового содержания йода в белковой матрице
Рентгенофлуоресцентный анализ (РФА)
Для определения элементов в молоке и молочных продуктах наиболее часто используются следующие методы: атомно-абсорбционный анализ (ААА) с пламенной или электротермической атомизацией элементов, масс-спектрометрия с индуктивно-связанной плазмой (МС-ИСП) и атомно-эмиссионная спектрометрия с индуктивно-связанной плазмой (АЭС-ИСП). Менее распространены нейтронно-активационный анализ (НАА), рентгеноспектральный анализ с протонным возбуждением (РСА П) и рентгенофлуоресцентный анализ (РФА). Среди других методов также можно отметить следующие: ААА с гидридным атомизатором и приставкой «холодного пара», атомно-флуоресцентный анализ (АФА), газо-жидкостную хроматографию в сочетании с МС-ИСП и АЭС-ИСП, потенциометрию.
Молоко и молочные продукты являются очень чувствительной системой к загрязнению в процессе пробоподготовки и анализа. Проблемы, возникающие при использовании какого-либо метода для определения элементов в молоке, обусловлены, в частности, сложным биоорганическим составом матрицы, очень низкими содержаниями многих микроэлементов в сочетании с высокими содержаниями щелочных и щелочноземельных металлов, хлора и фосфора. Большинство перечисленных выше методов ориентировано на анализ растворов после разложения органической матрицы молока и молочного продукта.
Этап разложения молока занимает около 61 % от времени, затраченного на анализ, и его вклад в суммарную погрешность анализа составляет примерно 30 %. В ряде случаев погрешность анализа, связанная с разложением, обусловлена присутствием молочного жира в молоке, который не всегда удается полностью перевести в раствор.
Среди недеструктивных методов следует отметить РФА [9,10], который позволяет анализировать высушенные или лиофилизованные образцы молочных продуктов без какой-либо химической обработки. Другими его достоинствами является многоэлементность, относительно короткое время пробоподготовки и измерения образца, эффективное определение летучих элементов, в частности, хлора и йода [Crecelius E.A.]. В ряде работ отмечено, что недостатками недеструктивного варианта РФА являются высокие пределы обнаружения, что затрудняет определение высокотоксичных элементов, таких как Cd, Hg, Pb и As.
Публикации, посвященные РФА молока [9], и решаемые в них задачи весьма немногочисленны, хотя во многих работах отмечаются большие возможности этого метода при определении элементов в молоке и молочных продуктах.
Целью данной части исследования являлось рассмотреть состояние и особенности РФА молочных йодированных белков, оценить возможность использования метода рентгено-флуоресцентного анализа для недеструктивного прямого определения валового содержания йода в йодированных молочных белках и продуктах, содержащих йод.
Измерения выполняли совместно на базе методического центра компании «Термо Техно» на энергодисперсионном анализаторе ARL QUANT’X. Thermo Fisher Scientific. В качестве матрицы для построения калибровочной прямой использовали крахмал картофельный ГОСТ 7699-78. Для приготовления контрольных точек с известным валовым содрежанием йода использовали йодат калия х.ч., точную навеску которого помещали в точно взвешенное количество крахмала, интенсивно перемешивали в течение 2-3 мин. на лабораторной мельнице.
Анализируемые пробы массой 2 г после тщательного измельчения в лабораторной мельнице прессовали под давлением 2 т. при одинаковых условиях в формах диметром d = 3.2 см.
Далее в соответствии с методикой измерения спектров РФА изучались пробы исходных йодированных белков «Биойод» и «Йодказеин».
Контроль анализируемого участка пробы осуществлялся с помощью CCD камеры с регулируемым размером рентгеновского пучка от 1 до 10 мм. и вращением пробы вокруг оси детектора (Si(Li) детектор с электрическим охлаждением).
Широкие возможности программы управления бесстандартного анализа UniQuantTM позволили оценить содержание йода в пробах недеструктивным способом.
Правильность результатов недеструктивного РФА молочных йодсодержащих продуктов оценивали с помощью стандартных образцов (добавки йодата калия к матрице крахмала) или с помощью контрольного метода (ИВА).

Рис.6. Схема образования флуоресцентного излучения от атомов йода после рентгеновского возбуждения
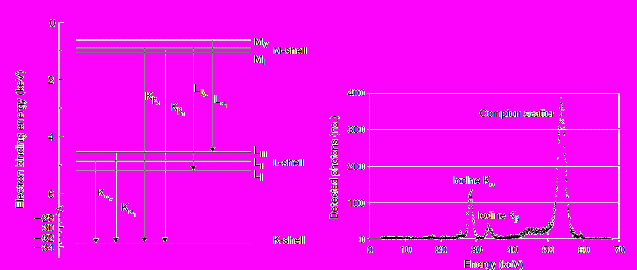
Рис.7. Спектры испускания флуоресцентного излучения от атомов йода после рентгеновского возбуждения [6].
Результаты валового содержания йода в анализируемых пробах представлены в сводной таблице раздела Приложение.
Суммарное валовое количество йода находится в интервале заявленного производителем содержания.