
Курс лекций 1999-2000 гг
| Вид материала | Курс лекций |
СодержаниеВероятность_внутри / вероятность_на_поверхности FEBS Letters |
- Курс лекций 1999-2000, 14768.78kb.
- Курс лекций для студентов заочного обучения Бурмистрова Л. А., Финансы предприятий:, 1991.45kb.
- Курс лекций. Спб, 1118.16kb.
- Курс лекций. Спб, 172.51kb.
- Курс лекций. Спб, 639.95kb.
- Цнж курс «Управление газетой», 1997 г.; «Триз-шанс» (Москва) курс «Приемы рекламы, 21.89kb.
- В. Б. Аксенов Краткий курс лекций, 1098.72kb.
- Курс лекций Барнаул 2001 удк 621. 385 Хмелев В. Н., Обложкина А. Д. Материаловедение, 1417.04kb.
- Основы политологии: Курс лекций. 2-е изд., доп. Ростов на/Дону.: Феникс, 1999. 573, 14.9kb.
- Г. И. Невельского Н. Н. Жеретинцева Курс лекции, 1964.49kb.
Лекция 19
С тех пор как стало понятно, что аминокислотная последовательность белковой цепи определяет ее пространственную структуру — возникла проблема предсказания этой структуры по последовательности аминокислотных остатков в белковой цепи.
Чем вызвана потребность в предсказании белковых структур, — кроме чисто интеллектуального интереса: удастся это сделать или нет? Тем, что экспериментально пространственную структуру белка определить куда труднее, чем его аминокислотную последовательность. А понимание механизма действия белка, подбор искусственных ингибиторов или активаторов к нему, — и часто даже просто определение того, чем он занимается в клетке, — настоятельно требует знания его пространственной структуры...
И — конечно же! — интерес к предсказанию пространственных структур белков подогревается воспоминанием о том, какое решающее значение имело предсказание строения двойной спирали ДНК для понимания всего генетического механизма.
Сейчас известно уже порядка сотни тысяч белковых последовательностей. Но "всего" для нескольких тысяч из них, т.е. всего для нескольких процентов определены, рентгеном или ЯМР, их пространственные структуры. При этом многие из недавно определенных последовательностей просто считаны с ДНК или РНК, т.е. никто не определил на опыте, чем занимаются сделанные из них белки.
Что же можно сказать о трехмерных структурах тех последовательностей (я их буду называть "новыми"), для которых эксперимент — рентген или ЯМР — еще не сказал своего слова?
Прежде всего возникает мысль о предсказании трехмерной структуры "новой" последовательности на основании родственного сходства — или, как говорят, "гомологии" ее первичной структуры с какими-то из "старых" последовательностей, пространственное строение коих уже расшифровано. Опыт показывает, что даже не очень сильного сходства последовательностей достаточно для очень хорошего сходства пространственных структур: как говорят, пространственная структура более консервативна, чем аминокислотная последовательность.
Установление гомологии первичных структур — действительно, очень мощный метод выяснения родства структур (причем не только белков, а и фрагментов ДНК и РНК — но я буду говорить о белках).
Однако надежно он работает, надежно устанавливает сходство первичных пространственных структур только на достаточно близких последовательностях. Этот случай иллюстрируется Рис.19-1.
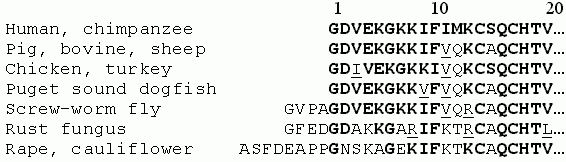
Рис.19-1. Гомологичные аминокислотные последовательности N-концевых фрагментов цитохромов c различных митохондрий и хлоропластов эукариотов. Жирным шрифтом выделены остатки, идентичные оным в человечьем (human) белке, подчеркнуты — сходные с ними. Выравнивание аминокислотных последовательностей взято из [6].
Хуже обстоит дело, когда белки в семействе сильно варьируют (Рис.19-2).
В этом случае на помощь приходит компьютер. Разработано множество программ, ищущих гомологии; с ними можно работать по Интернет. Назову только самые популярные из этих программ: ссылка скрыта и ссылка скрыта. Все они строят выравнивание (alignment) последовательностей, добиваясь наибольшего сходства между ними. При этом за повышение сходства часто приходится платить "разрывом" последовательностей (см. знаки "-" на Рис.19-2).
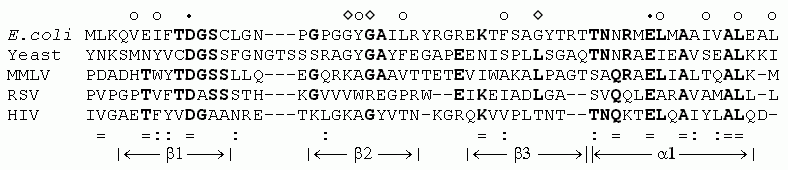
Рис.19-2. Аминокислотные последовательности N-концевых фрагментов рибонуклеаз Н бактерии (E.coli), эукариота (дрожжи, yeast), и трех разных вирусов. Множественное выравнивание делалось так, чтобы не допустить разрывов последовательностей (см. "- - -") внутри - и -структурных участков. Жирным шрифтом выделены остатки, идентичные в трех и более из этих пяти последовательностей. Черными точками отмечены остатки активного центра, пустыми кружками и ромбами — остатки, вовлеченные в два гидрофобных ядра этого белка. Внизу отмечены остатки, совпадающие ( = ) и сходные ( : ) у последовательностей из RSV и HIV (помещенных в двух нижних строках выравнивания), а также указана вторичная структура рассматриваемых белков. Картинка, с небольшими изменениями, взята из [7].
Разные программы по-разному оценивают, чего стоит совпадение остатков, чего — сходство, чего — несовпадение, чего — начало разрыва, чего — каждый дополнительный остаток в разрыве. Все эти оценки оптимизируются авторами так, чтобы удовлетворительно выделять белки, сходство которых уже известно из других данных, и потом "зашиваются" в программу. Пользуясь программой, люди ("пользователи") обычно даже не знают, что "хорошо" согласно этой программе, что "плохо", а просто говорят: "установлено, что гомология последовательностей составляет 25%" — имея в виду, что 25% выровненных остатков совпали друг с другом.
Встает вопрос — свидетельствуют ли эти 25% о сходстве последовательностей? Для ответа на этот вопрос необходимо сравнить, пользуясь той же программой, заведомо несходные последовательности. И тут выясняется, что "гомология" несходных белков (Рис.19-3) обычно составляет 10-15%, иногда — 20%, и порой — даже 25%!

Рис.19-3. Выравнивание аминокислотных последовательностей непохожих, негомологичных белков [в данном случае — -спирального РНК-связывающего белка (rop) и -структурного белка холодового шока (mjc)] часто дает 10-15% совпадающих аминокислотных остатков [в данном примере — 10 остатков (см. жирный шрифт) из 69, т.е. 14.5%]. Выравнивание сделано программой BLAST.
Эти цифры меняются от программы к программе. Однако накопленный опыт показывает, что тогда, когда "хорошая" (по общему мнению) программа дает совпадение свыше 30-35% остатков, — то выявленной гомологии можно смело доверять (с оговоркой: при длине сравниваемых последовательностей свыше 50, а лучше — 100 остатков). Правда, надо учитывать что 30 — 35% гомологии между последовательностями, верно (как правило) свидетельствуя об их родстве, позволяют правильно наложить друг на друга только 70-80% их пространственных структур, давая неверное предсказание о сходстве остальных 20-30%. А для того, чтобы верно проследить структуру 95% главной цепи "нового" белка, нужно, чтобы его гомология с белком с известной структурой достигала 40 — 50%.
Если же сходство пары последовательностей не превышает 10-15% — то их родство обычно нельзя обнаружить: такое сходство находится на уровне шума (что, однако, не является доказательством, что белки не похожи, не гомологичны — я к этому еще вернусь). А от 15 до 25 и даже до 30% простирается "сумеречная зона": кажется, что белки гомологичны, — но кто поручится?...
К сожалению, все эти цифры не вполне одинаковы у разных программ (и у разных режимов их работы), а к программе они, эти цифры, обычно не прилагаются (они есть в исходных статьях, но кто их читает...), — так что я бы рекомендовал, прежде чем доверяться любой такой программе, проверить ее (именно ее, и именно в используемом Вами режиме) на известных вам белках примерно той же длины (и сходных, и несходных) и понять, "что такое хорошо и что такое плохо" (другой вариант: прочесть исходную статью...).
Больше всего все эти оценки достоверности и недостоверности найденного сходства "плавают" от программы к программе из-за того, что разные авторы по-разному оценивают "штраф" за разрыв последовательности. Если его положить нулевым, то есть позволить делать любые разрывы "бесплатно", — случайно выбранные белковые (и вообще 20-буквенные) последовательности дают сходство на уровне 30-35% (а ДНКовые, 4-буквенные — на уровне 65%)!
Опыт показывает, что оптимальное отделение "похожих" от "непохожих" белковых последовательностей достигается, когда начало разрыва последовательности штрафуется в цену двух или трех дополнительных совпадений аминокислотных остатков, а за удлинение разрыва платится примерно 1/20 — 1/100 этой цены за каждый дополнительный остаток в разрыве.
Я умышленно не говорю ничего о математике, лежащей в основе алгоритмов поиска гомологий. Это нас увело бы слишком далеко. Хочу, однако, произнести ключевые слова: "динамическое программирование". Это — название самого мощного метода, применяемого для оптимизации одномерных систем (а последовательность — система именно одномерная), — в частности, для оптимизации выравнивания одной последовательности относительно другой.
Можно ли распознать гомологичность, родственность последовательностей, если их сходство лежит ниже уровня в 30%, — т.е. в "сумеречной зоне" или даже ниже ее? Можно — но для этого надо сравнивать интересующую нас последовательность со многими последовательностями семейства, и обращать внимание преимущественно на те позиции в цепи, что доказали свою консервативность именно в этом семействе.
Рисунок 19-2 показывает, что рибонуклеаза Н вируса иммунодефицита человека (HIV) имеет не очень высокое — "на уровне шума" — сходство с другими рибонуклеазами Н, если рассматривать всю цепь (так, из 60 выровненных остатков у нее есть всего 9 общих — 15% совпадений — с рибонуклеазой Н из RSV). Однако это сходство проявляется именно в тех ключевых районах, где все остальные рибонуклеазы похожи друг на друга. Это резко повышает достоверность такого сходства. А если еще учесть, что эти "ключевые районы" совпадений охватывают все аминокислотные остатки активного центра, и что сходство концентрируется в участках вторичной структуры, и что оно охватывает около 30% (а не 15%) остатков гидрофобных ядер белка, — высокая достоверность переходит в уверенность в правильном опознании гомологии.
Для опознания гомологии "новых" последовательностей также удобно пользоваться "консенсусными последовательностями" (Рис.19-4), выведенными для уже изученных белковых семейств и подчеркивающими их наиболее консервативные черты. Иногда такие консенсусные последовательности (снабженные данными по частотам встречаемости остатков в каждом месте цепи) называют "профилями первичных структур".
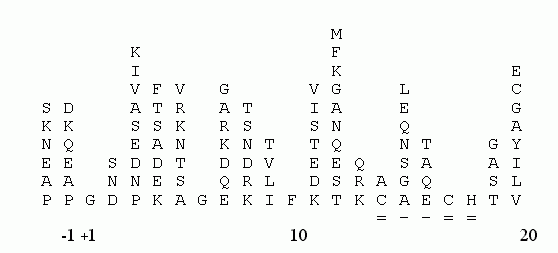
Рис.19-4. Аминокислотный состав различных позиций в N-концевых фрагментах цитохромов c митохондрий эукариот. Самые важные, консервативные остатки цепи определены однозначно. Подчеркнута последовательность "сайта" Cys-X-X-Cys-His, отвечающего за связывание гема в подавляющем большинстве цитохромов (и не только c, и не только эукариот). Выравнивание аминокислотных последовательностей взято из [6].
При опознавании функционального сходства белков следует также обращать внимание на уже установленные для многих функций "сайты" — более или менее короткие последовательности, обеспечивающие эти функции (см. на Рис.19-4 сайт Cys-X-X-Cys-His, связывающий гем в цитохромах). Такие сайты собраны в библиотеки, и их поиском занимаются специальные программы, из которых ссылка скрыта является наиболее популярной.
При установлении структуры "нового" белка по его гомологии с уже изученным надо ясно отдавать себе отчет, что сходство пространственных структур может не распространяться на районы, где последовательности сильно разошлись. В основном это (см. Рис.19-2) районы петель, нерегулярных конформаций белковой цепи. Здесь, с весьма переменным пока успехом, приходится прибегать к конформационным расчетам и другим методам гомологического моделирования, на которых я останавливаться не буду.
Перейдем к методам предсказания пространственной структуры "новых" последовательностей, не имеющих видимой гомологии с уже расшифрованными белками.
К ним относится около 2/3 "новых" последовательностей. Поэтому о пространственной укладке большинства последовательностей, получаемых в ходе выполнения генетических проектов, мы не можем догадаться по их гомологии с белками уже известными — она или слишком слаба для обнаружения, или отсутствует. Тут-то и возникает настоятельная потребность в решении задачи предсказания пространственной структуры — а, в перспективе, и функции белка, — по его аминокислотной последовательности (Рис.19-5).
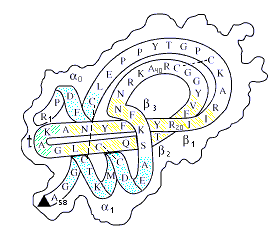
Рис.19-5. Схема первичной и пространственной структуры маленького белка (панкреатического ингибитора трипсина). Ход главной цепи изображен на фоне общего контура молекулы; выделены -спирали, -тяжи, резкий поворот цепи (t) и цистеиновые мостики ( - - - ). Так как белок сворачивается сам собой, то — в принципе — все это можно предсказать по одной лишь первичной структуре белка. Боковые группы здесь не показаны, но — в принципе — и их расположение в пространстве тоже можно предсказывать.
Надо сразу сказать, что абсолютно надежных и точных методов предсказания белковых структур сейчас нет.
Причин тому, видимо, две: (а) ограниченная точность энергетических оценок, на которых базируется теоретический расчет белковых структур, и (б) сравнительно малая разность между "правильно" и "неверно" уложенными белковыми цепями. Я хочу особо подчеркнуть последнее обстоятельство, — оно резко отличает ситуацию в белках (и РНК) от ситуации в ДНК, — к большому огорчению людей, занявшихся предсказаниями структур белков по их аминокислотным последовательностям. В ДНК комплементарное спаривание нуклеотидов наблюдается по всей длине двойной спирали, а оно обеспечивается тем, что одна цепь по своей первичной структуре строго комплементарна другой. Это и дает огромный свободно-энергетический выигрыш "правильной" структуры ДНК над всеми "неправильными". А в белках (и РНК) ни какой-либо строгой комплементарности, ни следующего из нее огромного энергетического преимущества "правильной" укладки цепи не наблюдается...
Однако существующие методы дают все более полную и надежную информацию о возможном строении (или, чаще, о возможных вариантах строения) белка или, особенно, его структурных элементов.
В прагматическом аспекте, вопрос о предсказании трехмерного строения белка сейчас ставится следующим образом: похожа ли пространственная структура рассматриваемой белковой последовательности на какую-либо из уже известных пространственных белковых структур? Если да, то как рассматриваемая последовательность вписывается в эту структуру? Если нет, можем ли мы указать какие-либо характерные детали пространственной организации рассматриваемой последовательности?
Я знаю несколько случаев, когда ответы на эти вопросы существенно способствовали экспериментальному определению структуры и/или функции изучаемой аминокислотной последовательности, помогли планировать белково-инженерные эксперименты и так далее. Впрочем, должен сказать, что я бы с большим интересом прочел бы хороший обзор о практическом применении предсказаний белковых структур...
Перейдем теперь к методам предсказания белковых структур. Рассматривая эти методы, я буду уделять особое внимание тем идеям, и в основном физическим идеям, что лежат в их основе.
Есть две стратегии предсказания белковых структур. Согласно первой стратегии, белковая структура ищется как результат кинетического процесса сворачивания.
Согласно второй, — она ищется как структура с минимально возможной для данной цепи свободной энергией.
В принципе, по-видимому, обе эти стратегии могут привести к правильному результату, так как самая стабильная структура белковой цепи, несмотря на опасения Левинталя, образуется достаточно быстро (этому вопросу была посвящена отдельная лекция, и сейчас я позволю себе его не касаться).
Важно, однако, что, рассматривая предсказание белковых структур, мы можем отвлечься от путей сворачивания и рассматривать только результат — стабильную структуру белка. Тем более, что первая стратегия — стратегия имитационного моделирования процесса сворачивания белка — пока существенного прогресса не дала: уж очень сложны все расчеты, и к тому же — все же весьма приблизительны все потенциалы взаимодействий. Вторая стратегия оказалась более успешной.
Начнем с определения стабильной вторичной структуры белка по его аминокислотной последовательности. -спирали и вытянутые -участки — важнейшие элементы белковых глобул, во многом определяющие, как вы помните, их общую архитектуру (Рис.19-5).
Забудем пока о том, что белковая цепь свернута в глобулу, т.е. рассмотрим развернутую белковую цепь. Можем ли мы предсказать ее вторичную структуру по аминокислотной последовательности? Оказывается — да, в основном да, пусть и не абсолютно точно.
Прежде всего, — какие аминокислотные остатки будут стабилизировать вторичную структуру — например, -спираль, — а какие будут разрушать ее?
Эксперименты дают прямой ответ на этот вопрос. Я имею в виду огромную работу по оценке - и -образующих способностей аминокислотных остатков, проведенную в группах Шераги, Фасмана, Болдвина, Фершта, Серрано, ДеГрадо, Кима и в ряде других групп (и, в частности, у нас — В.Е.Бычковой и О.Б.Птицыным). Кроме того, богатый (и хорошо совпадающий с физико-химическим экспериментом) материал дает статистика аминокислотных последовательностей - и -структур в белках. Рисунок 19-6 суммирует важнейшие (те, которые стоит запомнить) из оценок, полученных всеми этими методами.
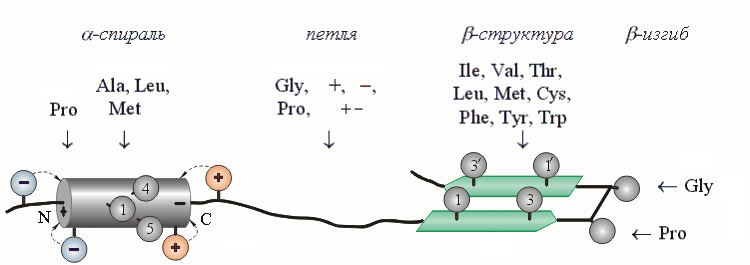
Рис.19-6. Шаблоны -спирали, петли, -структуры и -изгиба — те остатки, которые стабилизируют их или их отдельные части. "+" означает все положительно заряженные аминокислоты, "" — все отрицательно заряженные, "+ " — все аминокислоты с диполем в боковой цепи. Показан также стабилизирующий - и -структуру порядок чередования гидрофобных групп в цепи (см. нумерованные группы). Такого типа чередование приводит также к образованию гидрофобных и полярных поверхностей -спиралей и -тяжей.
Здесь надо, однако, сразу сказать, что все закономерности, наблюдающиеся в структурах белков, носят вероятностный, частотный характер. Несмотря на многочисленные попытки, не удалось выделить никакого четкого "кода" белковых структур — то есть ничего похожего на то четкое A-T и G-C спаривание нуклеотидов, которое свойственно двойной спирали ДНК. Впрочем, надо признать, что уже в РНК — с их разнообразным, в отличие от ДНК, репертуаром пространственных структур — спаривание нуклеотидов происходит далеко не столь однозначно...
Подавляющее большинство из полученных экспериментальных и статистических оценок можно легко понять, исходя из физики и стереохимии аминокислот. Мы уже говорили об этом на одной из прошлых лекций.
Так, Pro не любит входить ни в -спираль (кроме ее N-концевого витка), ни в -структуру. Почему? Потому, что у него нет NH-группы, и он не может завязывать соответствующие - и -структуре водородные связи; а на N-конце спирали NH-группа таких связей и не должна завязывать, — и Pro там встречается часто, тем более что его угол уже фиксирован пролиновым кольцом в подходящем положении. По тем же причинам часто встречается Pro и на N-конце изгибов.
А вот Ala стабилизирует -спираль, — а Gly разрушает и ее, и -структуру, и способствует образованию клубка. С чем связана эта разница? С тем, что область конформаций, т.е. область допустимых углов для Gly в клубке гораздо больше, чем для Ala, — в то время как допустимые конформации для обоих этих остатков в -спирали примерно совпадают.
По аналогичным причинам разветвленные остатки — Val, Ile и Thr — больше стабилизируют -структуру, где их боковые группы имеют 3 разрешенных поворотных изомера, чем -спираль или клубок, где они имеют только 1 изомер (при каждом значении и ).
Вообще же гидрофобные группы склонны несколько чаше входить в - и -структуру, где они могут слипаться в гидрофобных кластерах (см. Рис.19-6), чем в клубок, где они этого делать не могут. А вот боковые группы с диполями, особенно — в короткой боковой цепи, больше склонны входить в нерегулярные участки цепи, где они могут завязывать дополнительные нерегулярные водородные связи, чем в - и -структуры, где доноры и акцепторы водородных связей уже насыщены внутрицепными связями.
Более того, влияние аминокислотных остатков на вторичную структуру можно оценить теоретически. Так, еще до получения экспериментальных оценок, в начале и середине 70-х годов, мы с О.Б.Птицыным предсказали, что отрицательно заряженные остатки должны стабилизировать N-конец спирали, притягиваясь к его положительному парциальному заряду, и дестабилизировать ее С-конец, отталкиваясь от отрицательного парциального заряда спирального диполя. Положительно же заряженные остатки должны действовать в прямо противоположном направлении. При этом потенциал каждого такого взаимодействия должен, по теоретической оценке, составлять 1/4 или 1/3 килокалории. Что и было подтверждено экспериментально.
Зная, какие аминокислотные остатки стабилизируют середину спирали, какие — ее N-конец, а какие — C-конец, мы получаем нечто вроде "шаблона" спирали. Шаблон -спирали можно описать так: если начало фрагмента белковой цепи обогащено отрицательно заряженными группами, да еще там стоит Pro; если середина этого фрагмента обогащена остатками Ala, Leu и Met, а Pro и Gly там нет; и если его С-конец содержит положительно заряженные боковые группы, — то перед нами -спиральный участок. Кроме того, для стабильности -спирали полезен, а для ее включения в глобулу — просто необходим определенный (Рис.19-6) порядок чередования гидрофобных групп в цепи: этот порядок способствует слипанию этих групп и, кроме того, приводит к образованию на спирали сплошной гидрофобной поверхности, необходимой для ее прилипания к глобуле.
То есть "шаблон" качественно описывает аминокислотную последовательность, подходящую для образования -спирали. Чем лучше аминокислотная последовательность удовлетворяет этому шаблону — тем вероятнее спираль в данном месте цепи. Такое же описание — "шаблон" — можно составить и для участков, пригодных для образования -структуры. А также — для участков, особо пригодных для образования -изгибов и петель.
Более того, "шаблоны" можно применять и для описания кусков цепи, образующих более сложные структуры, — например, для описания суперспиралей, состоящих из двух параллельных -участков и -спирали между ними (Рис.19-7). Такие структуры типичны для доменов, связывающих нуклеотиды. Особо важную роль в "шаблоне" играют так называемые "ключевые позиции", которые могут быть заняты только строго определенными аминокислотными остатками, — например, Gly: только этот остаток может находиться в конформации с >60о, недоступной всем остальным остаткам.
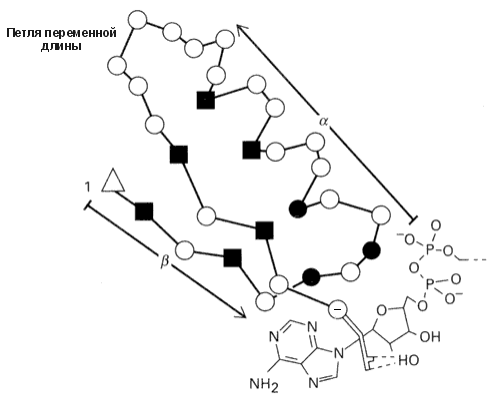
Рис.19-7. "Шаблон" суперспирали , связывающей нуклеотиды. Квадратики отмечают ключевые позиции, обычно занимаемые сравнительно небольшими гидрофобными остатками (Ala, Ile, Leu, Val, Met, Cys): это — гидрофобное ядро суперспирали . Черные кружки — позиции, занимаемые только Gly: здесь находятся резкие повороты цепи. Пустой треугольник отмечает первую позицию мотива , где обычно находится положительно заряженная или дипольная боковая цепь. В последней (-) позиции мотива находится аминокислота Asp или Glu, связывающая лиганд (нуклеотид). Рисунок, с небольшими изменениями, взят из R.K.Wierenga et al., J. Mol. Biol. (1986) 187:101-107.
Но вернемся к расчету вторичной структуры. Зная вклады отдельных взаимодействий в стабильность -спирали, мы можем рассчитать свободную энергию спирализации любого участка цепи, а следовательно — и Больцмановскую вероятность образования спирали в любом месте полипептидной цепи, еще не свернувшейся в глобулу. Суммируя и усредняя эти вероятности, мы можем рассчитать и среднюю спиральность такого "несвернутого" полипептида. Вот уже более 15 лет мы используем для этого нашу программу ALB. В одном из режимов ("unfolded chain" — "развернутая цепь") она позволяет рассчитывать содержания - и -структуры в полипептидах и в несвернутых белковых цепях, — причем при разной температуре, ионной силе и рН раствора. Потом результат можно сравнить с опытными данными — например, с КД спектрами. Рисунок 19-8 показывает, что теоретический расчет неплохо совпадает с опытом.
-
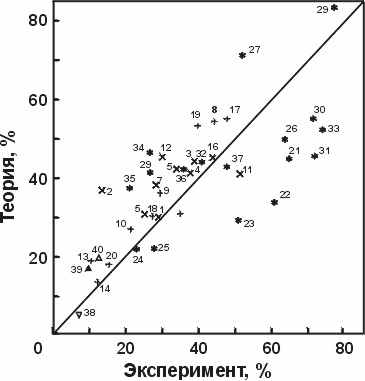
Рис.19-8. Теоретическая (вычисленная программой ALB — unfolded chain) и экспериментально найденная спиральность нескольких десятков пептидов при температуре 0о-5оС и разной ионной силе и рН раствора. Рисунок взят из A.V.Finkelstein, A.Y.Badretdinov & O.B.Ptitsyn, Proteins (1990) 10:287-299.
Переходя к расчету и предсказанию вторичной структуры белков, глобулярных белков, необходимо учесть, что здесь к взаимодействиям, существующим в несвернутых цепях, добавляется взаимодействие каждого участка цепи с глобулой, строения которой мы не знаем. Точнее, мы не знаем ее детального строения, но знаем, что участки цепи как-то примыкают к гидрофобному ядру белка. В простейшем приближении взаимодействие с ядром можно аппроксимировать взаимодействием с "гидрофобным озером", на котором плавает белковая цепь (Рис.19-9).
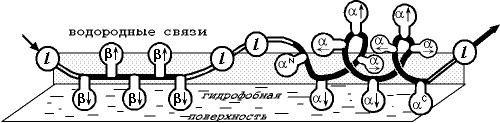
Рис.19-9. Флуктуирующая вторичная структура белковой цепи (здесь: — -тяж, l — петля, — -спираль) на поверхности гидрофобного озера, имитирующего белковую глобулу ("модель плавающих бревен"). Модель учитывает разное чередование обращенных к поверхности озера (
 ), от поверхности (
), от поверхности ( ) и вдоль нее () боковых групп в разных вторичных структурах, а также эффекты на N- и С-концах спирали, объединенные и приписанные ее, соответственно, N- и С-концевым остаткам (N ,C). Рисунок, с небольшими изменениями, взят из O.B.Ptitsyn & A.V.Finkelstein, Bipolymers (1983) 22:15-25.
) и вдоль нее () боковых групп в разных вторичных структурах, а также эффекты на N- и С-концах спирали, объединенные и приписанные ее, соответственно, N- и С-концевым остаткам (N ,C). Рисунок, с небольшими изменениями, взят из O.B.Ptitsyn & A.V.Finkelstein, Bipolymers (1983) 22:15-25. Зная из опыта силу гидрофобных взаимодействий, а из стереохимии - и -структуры — мотивы чередования в цепи боковых групп, глядящих в одну и ту же сторону и способных, следовательно, одновременно взаимодействовать с гидрофобной поверхностью, — мы можем сосчитать вероятность образования -спирали и -структуры в каждом месте белковой цепи. Этим также — но уже в режиме "глобулярная цепь" ("globular chain") — также занимается программа ссылка скрыта (кстати, к ней можно обращаться по Интернет: serpukhov.su/~rykunov/alb/).
Вероятности рассчитываются при комнатной температуре. Почему при комнатной? Не лучше ли выделять только самое лучшее по энергии расположение - и -структур в цепи, т.е. рассчитывать все вероятности при 0оК?
Прежде всего, конечно, расчет относится к комнатной температуре потому, что все экспериментальные оценки стабильности, на которых базируется расчет, относятся к этой температуре.
Но еще важнее то, что вероятности, рассчитанные именно при такой температуре (300оК), лежащей чуть ниже нормальной температуры плавления белка (350оК) наилучшим образом позволяют отделить более вероятные - и -участки, в которых мы можем быть более или менее уверены, от тех "менее вероятных", в которых мы уверенными быть не можем.
Ведь, в сущности, мы стараемся предсказать вторичную структуру белка только по части тех взаимодействий, что ее держат в действительности: мы знаем (и то приблизительно) внутренние взаимодействия в этой структуре, но не знаем (или можем лишь крайне грубо оценить) те, что действуют между рассматриваемым куском цепи и остальной глобулой. А они очень мощны.
То есть мы находимся в том же положении, как если бы пытались предсказать, будет ли данный остаток внутри белка или на его поверхности, зная только его собственную гидрофобность — и больше ничего. И мы знаем, что такая задача имеет ответ, — но ответ не точный, а вероятностный. Этот ответ содержится в наблюдаемой статистике распределения остатков между нутром и поверхностью глобулы. Мы уже знаем, что она выглядит так:
-
Содержание