К. Б. Терёшкина молекулярная динамика белков и пептидов методическое пособие
| Вид материала | Методическое пособие |
СодержаниеК.В. Шайтан, С.С. Сарайкин, Метод молекулярной динамики, 1999, n.ru/library/md/default.php Расчёт ньютоновских траекторий движения. |
- Тема: Аминокислоты, пептиды, белки, 124.2kb.
- План лекций по биологической химии для студентов лечебного факультета, 66.35kb.
- Влияние экспрессии гетерологичных генов хитинсвязывающих белков и гевеин-подобных антимикробных, 1284.68kb.
- 2004 статьи отечественные журналы, 552.71kb.
- В. А. Жернов апитерапия учебно-методическое пособие, 443.6kb.
- На базе научно-практического центра эндоваскулярной нейрорентгенохирургии амн украины, 491.31kb.
- Тема обмен белков. Вопросы лекции, 90.92kb.
- Биосинтез белков Интегрированный урок в 10-м классе(химия и биология). Цель урока, 27.63kb.
- Рабочая программа дисциплины «биология клетки» (молекулярная биология) Код дисциплины, 225.32kb.
- В. Х. Хавинсон пептидная регуляция старения санкт-петербург «наука» 2009, 400kb.
Литература.
- M.P.Allen and D.J.Tildesley, Computer Simulation of Liquids, Oxford: Clarendon Press, 2002.
- Э.Э.Шноль, А.Г. Гривцов и.др., Метод молекулярной динамики в физической химии, М.: Наука, 1996.
- В.Г.Дашевский, Конформации органических молекул, М.: Химия, 1974.
- С.Мидзусима, Строение молекул и внутреннее вращение, M.: Изд-во иностр.лит., 1957.
- О.М.Полторак, Термодинамика в физической химии, М.: Высш. шк., 1991.
- И.Г.Каплан, Введение в теорию межмолекулярных взаимодействий, М.: Наука, 1982.
- Э.А.Мелвин-Хьюз, Физическая химия, М.: ИЛ, 1962.
- A.S.Lemak and N.K.Balabaev, A comparison between collisional dynamics and Brownian dynamics, // Molecular Simulation, 1995,15, 223-231.
- A.S.Lemak and N.K.Balabaev, Molecular dynamics simulation of polymer chain in solution by collisional dynamics method, // J.Comput.Chem., 1996,17, 1685-1695.
- L.D.Landau and E.Teller, On the theory of sound dispersion, // Physik.Zeits.Sowjetunion, 1936,10, 34-43.
- В.Л.Голо, К.В.Шайтан, Динамический аттрактор в термостате Берендсена и медленная динамика биомакромолекул, // Биофизика, 2002, 47 (4), 611-617.
- Х.Вестерхофф, К. ван Дам, Термодинамика и регуляция превращений свободной энергии в биосистемах, М.: Мир, 1992.
- Л.С.Полак, М.Я.Гольденберг, А.А.Левицкий, Вычислительные методы в химической кинетике, М.: Наука, 1984.
- К.В.Шайтан, А.А.Беляков, К.М.Леонтьев, С.С.Сарайкин, М.Г.Михайлюк, К.Б.Егорова, М.В.Орлов, Геометрия энергетической поверхности и конформационная динамика: от углеводородов - к белкам и пептидам, // Хим.физ., 2003, 22 (2), 57-68.
- Х.Аренс, Ю.Лёйтер, Многомерный дисперсионный анализ, М.: Финансы и статистика, 1985.
- Д.Худсон, Статистика для физиков, М.: Мир, 1970.
- Г.Шеффе, Дисперсионный анализ, М.: Наука, 1980.
- Н.Ф.Степанов, В.И.Пупышев, Квантовая механика молекул и квантовая химия, М.: МГУ, 1991.
- Н.Ф.Степанов, Квантовая механика и квантовая химия, М.: Мир, 2001.
- К.В.Шайтан, А.В.Немухин, Д.А.Фирсов, Т.В.Богдан, И.А.Тополь, Электронно-конформационные взаимодействия и значение эффективных зарядов на атомах в пептидах, // Мол.биол., 1997, 31, 109-117.
- Ю.А.Овчинников, Биоорганическая химия, М.: Просвещение, 1987.
- А.В.Финкельштейн, О.Б.Птицын, Физика белка: Курс лекций с цветными и стереоскопическими иллюстрациями, М.: Книжный дом "Университет", 2002.
Дополнительная литература.
- C.L.Brooks, M.Karplus, and B.M.Pettit, Proteins: A theoretical perspective of dynamics, structure and thermodynamics. In: Advances in chemical physics. V.LXXI (ed I.Prigogine & S.A.Rice). N.-Y.,1987.
- J.A.McCammon and S.C.Harvey // Dynamics of Proteins and Nucleic Acids. C.: Cambr. Univ. Press, 1987.
- T.Noguti, and N.Go // Proteins: Structure, Function, and Genetics, 1989, 5, 97-138.
- K.V.Shaitan, Protein dynamics and new approaches to the molecular mechanisms of protein functioning. In: Stochastic Dynamics of Reacting Biomolecules (ed. W. Ebeling, Yu. Romanovsky, L. Schimansky-Geier). World Scientific, 2003, 283-308.
- K.B.Egorova, K.V.Shaitan, and A.Yu.Ermilov, Molecular Dynamics of Strained Retinal in Various Electronic States. // IJQC, 2004, 96, 219-225.
- К.В.Шайтан, М.Г.Михайлюк., К.М.Леонтьев, С.С.Сарайкин, А.А.Беляков, Влияние дисульфидных связей на динамику лизоцима. // Биофизика 2003, 48, 2, 210-216.
- К.В. Шайтан, А.А. Беляков, Молекулярная динамика олигопептидов. 4. Динамические особенности часто и редко встречающихся дипептидных фрагментов белков // Биофизика 2002, 47, 2, 219-227.
- К.В.Шайтан, М.Г.Михайлюк., К.М.Леонтьев, С.С.Сарайкин, А.А.Беляков, Молекулярная динамика изгибных флуктуаций элементов вторичной структуры белков. // Биофизика 2002, 47, 411-419.
- А.Г.Ивайкина, Н.К.Балабаев, К.В.Шайтан, Определение вклада пептидной cтруктуры в энергетику реакций окисления-восстановления белков, содержащих Fe4S4 кластеры с помощью компьютерных МД экспериментов. // Биофизика, 2001, 46, 589.
- К.В.Шайтан, А.Я.Муковский, А.А.Беляков, С.С.Сарайкин, Статистические распределения дипептидов в белковых структурах и динамические свойства некоторых белковых фрагментов. // Биофизика, 2000, 45, 399-406.
- К.В.Шайтан, С.С.Сарайкин, Влияние амплитуды флуктуаций на коэффициент трения броуновского осциллятора в водной среде. // Биофизика, 2000, 45, 407-413.
- К.В.Шайтан, С.С.Сарайкин, А.К.Васильев, М.Г.Михайлюк, О влиянии электронного строения и динамических свойств радиопротекторов на их биологическую активность. // Биофизика, 1999, 44, 668-675.
- K.V. Shaitan, M.D. Ermolaeva, and S.S. Saraikin, Nonlinear dynamics of the molecular systems and the correlations of internal motions in the oligopeptides. // Ferroelectrics, 2000, 220, 205 220.
- К.В.Шайтан, П.П.Пустошилов, Молекулярная динамика монослоя стеариновой кислоты. // Биофизика, 1999, 44, 436-441.
- К.В.Шайтан, М.Д.Ермолаева, С.С.Сарайкин, Молекулярная динамика олигопептидов. 3. Карты уровней свободной энергии модифицированных дипептидов и динамические корреляции в аминокислотных остатках. // Биофизика, 1999, 44, 18-21.
- К.В. Шайтан, М.Д. Ермолаева, Н.К. Балабаев, А.С. Лемак, М.В. Орлов, Молекулярная динамика олигопептидов. 2. Корреляционные функции внутренних степеней свободы модифицированных дипептидов. // 1997, 42, 3, 558-565.
- К.В.Шайтан, Н.К.Балабаев, А.С.Лемак, М.Д.Ермолаева, А.Г.Ивайкина, О.С.Кислюк, М.В.Орлов, Е.В.Гельфанд, Молекулярная динамика олигопептидов. 1. Использование длинных траекторий и высоких температур для определения статистического веса конформационных подсостояний. // Биофизика, 1997, 42, 47-53.
- Н.К.Балабаев, А.С.Лемак, К.В.Шайтан, Молекулярная динамика и электронно-конформационные взаимодействия в ферредоксине. // Мол.биол., 1996, 30, 1348-1356.
- К.В.Шайтан, Динамика электронно-конформационных переходов в белках и физические механизмы функционирования биомакромолекул. // Мол.биол.,1992, 26, 264-284.
- К.В.Шайтан, Динамика электронно-конформационных переходов и новые подходы к физическим механизмам функционирования биомакромолекул. // Биофизика, 1994, 39, 949-967.
- К.В.Шайтан, Физические механизмы конформационной подвижности биополимеров. // В сб.: Равновесная динамика структуры биополимеров, Пущино, 1990, 9-19.
- A.S.Lemak, Collisional dynamics for molecules with constraints. Preprint NCBI, Pushchino, 1992.
- A.S.Lemak and N.K Balabaev, On the Berendsen thermostat. // Molecular Simulation, 1994, 13, 177-187.
- Н.К.Балабаев, А.А.Нурисламов, И.В.Упоров, К.В.Шайтан, Молекулярная динамика олигопептидов и их структурно-функциональная организация. // В cб.: Математические и вычислительные методы в биологии. Биомолекулярные системы. Пущино, 1987, 3-11.
- К.В.Шайтан, Энергетическая поверхность и конформационная динамика молекул. // Электрохимия, 2003, 39, 2, 212-219.
- N.K.Balabaev, A.A.Darinskii, I.M.Neelov, A.Zarembo, and F.Sundholm Computer Simulation of a Liquid-Crystal System of Semirigid Rodlike Linear Molecules // Polymer Science, Ser. A, 2002, 44, 11, 1146-1154.
- А.А.Берлин, М.А.Мазо, Н.К.Балабаев, Ю.К.Товбин. Молекулярно-динамическое поведение одно- и двухкомпонентных систем в узких щелевидных порах. // Химическая физика, 2002, 21,2, 3-8.
- В.В.Корнилов, А.Л.Рабинович, Н.К.Балабаев, Исследование структуры и свойств полиненасыщенных липидных монослоев методом молекулярной динамики // Журнал физической химии. 2002, 76, 11, 2018-2022.
- Н.К.Балабаев, А.А.Даринский, И.М.Неелов, Н.В.Лукашева, И.Эмри, Молекулярно-динамическое моделирование двухмерного полимерного расплава. // Высокомолек. соед., Сер. А, 2002, 44, 7, 1228-1239.
- А.А.Берлин, О.В.Гендельман, М.А.Мазо, Л.И.Маневич, Н.К.Балабаев, Плавление кристаллов из упругих и леннард-джонсовых сферических частиц. // Доклады АН, 2002, 382, 6, 798-801.
- ссылка скрыта
Молекулярная динамика.
Метод молекулярной динамики позволяет моделировать детальную микроскопическую картину внутренней подвижности макромолекулы. В его основе лежит расчет классических (ньютоновских) траекторий движения макромолекулы в фазовом пространстве координат и импульсов ее атомов, когда молекула рассматривается как система взаимодействующих классических частиц. Метод молекулярной динамики успешно используется в теоретических исследованиях структуры и динамики биологических макромолекул, жидкостей, газов и других молекулярных систем.
Ниже мы рассмотрим физико-математический аппарат, лежащий в основе группы методов, объединённых этим названием.
Здесь также приводятся примеры молекулярного кино, созданного с помощью этих методов. Молекулярное кино, это отображение молекулярной конформационной подвижности в реальном времени. Приведённые далее примеры иллюстрируют "жизнь молекул" в диапазонах от 0,5 до 10 пикосекунд
Расчёт ньютоновских траекторий движения.
В методе молекулярной динамики рассчитываются классические (ньютоновские) траектории движения атомов макромолекулы в силовом поле эмпирического атом-атомного потенциала, т. е. моделируется детальная микроскопическая картина внутренней тепловой подвижности макромолекулы в субнаносекундных интервалах времен. Основу метода составляет численное решение классических уравнений Ньютона для системы взаимодействующих частиц:

где
 - радиус-вектор i-го атома,
- радиус-вектор i-го атома,  - его масса,
- его масса,  суммарная сила, действующая на i-ый атом со стороны остальных частиц:
суммарная сила, действующая на i-ый атом со стороны остальных частиц: 
Здесь:
 -потенциальная энергия, зависящая от взаимного расположения всех атомов; n - число атомов.
-потенциальная энергия, зависящая от взаимного расположения всех атомов; n - число атомов.Задав координаты и скорости всех частиц в начальный момент времени, числено решают уравнения движения, вычисляя на каждом шаге все силы и новые координаты и скорости частиц. Температура определяется как средняя кинетическая энергия, приходящаяся на одну степень свободы системы:

Здесь N - полное число степеней свободы молекулы,
 - постоянная Больцмана. В случае изолированной системы N=3n-6, поскольку сохраняется ее полный импульс и момент импульса. Кроме того, в этом случае сохраняется полная энергия системы, а температура получается усреднением ее мгновенных значений T(t) по некоторому интервалу времени.
- постоянная Больцмана. В случае изолированной системы N=3n-6, поскольку сохраняется ее полный импульс и момент импульса. Кроме того, в этом случае сохраняется полная энергия системы, а температура получается усреднением ее мгновенных значений T(t) по некоторому интервалу времени. Потенциальная энергия молекулы задается в виде:

 +
+ +
+ +
+ +
+ +
+ +
+
где слагаемые отвечают следующим типам взаимодействий:
- химическим связям; - валентным углам; - торсионным углам; - плоским группам; - ван-дер-ваальсовым контактам; - электростатике; - водородным связям. Основные типы структурных взаимодействий представлены на рисунке: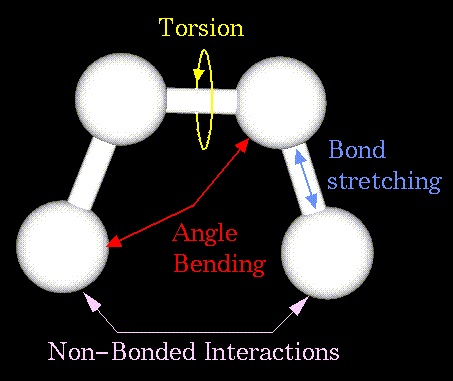
Указанные слагаемые имеют различный функциональный вид. Валентные длины поддерживаются за счет потенциала:

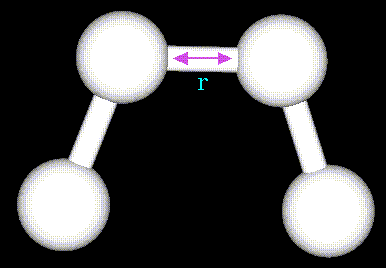
где суммирование проводится по всем химическим связям,
 - обозначение для равновесных валентных длин, r - текущие длины связей,
- обозначение для равновесных валентных длин, r - текущие длины связей,  - соответствующие силовые константы ссылка скрыта. Уравнение, описывающее потенциал валентных связей следует из закона Гука. Это уравнение параболы:
- соответствующие силовые константы ссылка скрыта. Уравнение, описывающее потенциал валентных связей следует из закона Гука. Это уравнение параболы: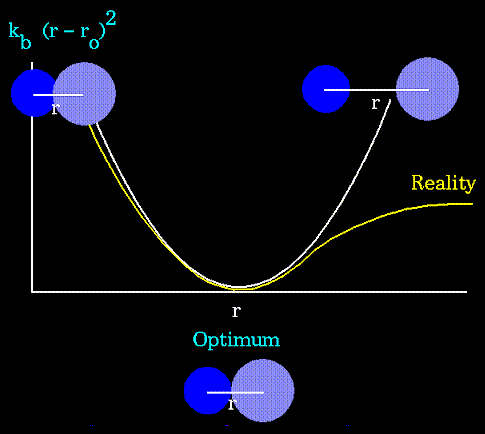
Валентные углы задаются потенциалами

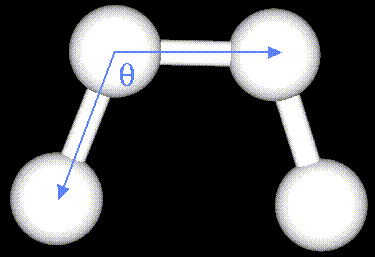
где
 - равновесные значения углов,
- равновесные значения углов,  - их текущие значения,
- их текущие значения,  - силовые константы ссылка скрыта. Уравнение, описывающее потенциал валентных связей также следует из закона Гука. Это уравнение параболы:
- силовые константы ссылка скрыта. Уравнение, описывающее потенциал валентных связей также следует из закона Гука. Это уравнение параболы: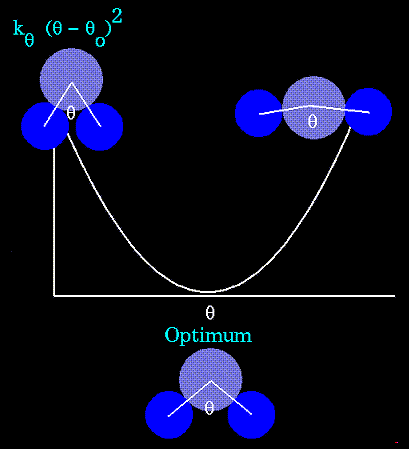
Энергия торсионных взаимодействий и потенциалов, отвечающих плоским группам, записываются в одинаковом виде:

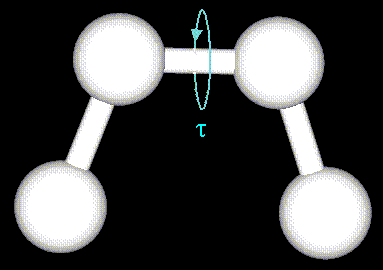
где n - кратность торсионного барьера,
 - сдвиг фазы, константы
- сдвиг фазы, константы  определяют высоты потенциальных барьеров двугранных углов
определяют высоты потенциальных барьеров двугранных углов  ссылка скрыта. Энергия торсионных углов может быть задана с помощью простой периодической функции, как это показано на рисунке:
ссылка скрыта. Энергия торсионных углов может быть задана с помощью простой периодической функции, как это показано на рисунке: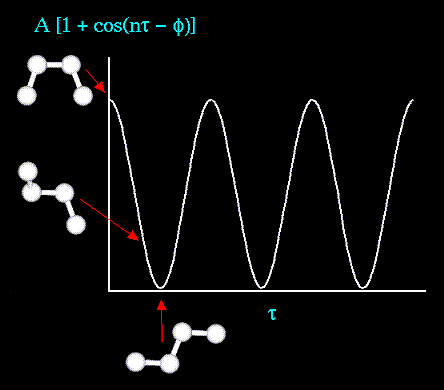
Ван-дер-ваальсовые взаимодействия атомов, разделенных тремя и более валентными связями описываются с помощью потенциалов Леннард-Джонса:

Параметры потенциала A и B зависят от типов атомов i и j, участвующих во взаимодействии;
 - расстояние между этими атомами. Аналитический вид такого потенциала представлен на рисунке:
- расстояние между этими атомами. Аналитический вид такого потенциала представлен на рисунке: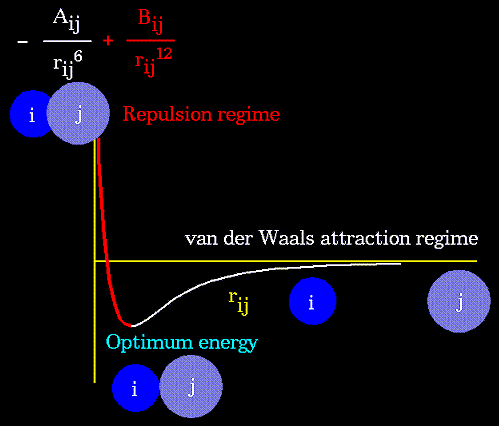
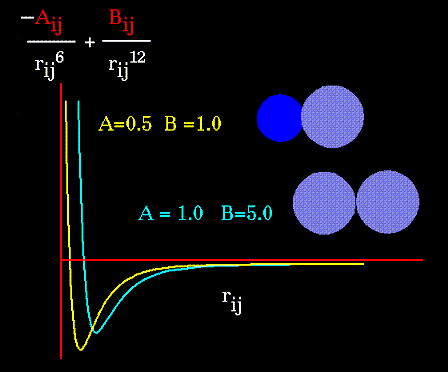
Очевидно, что вид потенциала зависит от свойств атомов, участвующих в образовании Ван-дер-Ваальсовых связей. Ниже, на рисунке представлен вид потенциала для разных пар атомов:
Электростатические взаимодействия задаются кулоновским потенциалом

где
 ,
,  - парциальные заряды на атомах,
- парциальные заряды на атомах,  - диэлектрическая проницаемость среды.
- диэлектрическая проницаемость среды.Водородные связи возникают и исчезают в процессе движения атомов между теми из них, которые имеют донорно-акцепторный статус. Функциональный вид потенциала водородной связи аналогичен потенциалу ван-дер-ваальсовым взаимодействий:

Существуют различные наборы параметров для потенциалов взаимодействий. Их значения определяются из учета различных типов экспериментальных данных (спектральные, калориметрические, кристаллографические) и квантовомеханических расчетов.
Литература:
- McCammon J.A., Harvey S.C., Dynamics of proteins and nucleic acids, Cambridge: Cambridge University Press, 1987.
- N.K. Balabaev, A.S. Lemak, Molecular dynamics simulation of ferredoxin in different electronic states. In: Laser Spectroscopy of Biomolecules, E.I. Korppi-Tommola, Ed., Proc. SPIE 1921, 375-385 (1993).
- Карплус М., Мак-Каммон Дж.Э. Динамика белковой структуры. В мире науки. 1986 №6 С.4-15
- Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S., Karplus M. CHARMM: A program for macromolecular energy minimization, and dynamics calculations. J/Comput. Chemistry. 1983. V.4. No.2. P.187-217.
- Mazur A.K., Abagyan R.A. New methodology for computer-aided modelling of biomolecular structure and dynamics. Non-cyclic structures. J. Biomol. Struct. Dyn. 1989. V.6. P. 815-832
Методы ускорения расчётов молекулярной динамики.
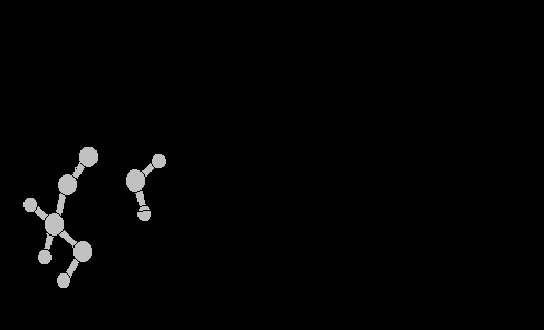
Время, необходимое для расчета траектории молекулы, можно значительно сократить, уменьшая число степеней свободы. Существует два способа ограничения движений длин валентных связей и углов. В одном случае длины валентных связей и значения валентных углов жестко фиксированы, в другом случае на них накладываются упругие ограничения с очень большой константой упругости. Статистические свойства жестко и упруго ограниченных систем, вообще говоря, различны. При разных формах упругого потенциала получаются статистически разные результаты, один из таких потенциалов соответствует жестко фиксированным валентным связям и углам. При этом, в общем случае, упругие потенциалы статистически предпочтительнее как для валентных связей, так и для валентных углов. Рассмотрим, например, следующий численный эксперимент: с помощью метода молекулярной динамики моделировали движение трехатомной (рис., а) и четырехатомной молекул (рис., б) в растворе со сферическими молекулами. Оказалось, что в случае жестких ограничений, в отличие от упругих, вектора, соединяющие первый и третий атомы в случае (а) и первый и четвертый атомы в случае (б), неравномерно распределены по сфере:
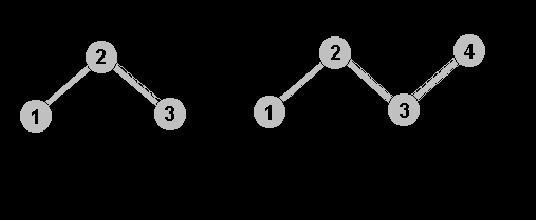
Последние исследования в этой области показали, что колебания валентных углов, по-видимому, связаны с коллективными движениями в молекуле и, из-за плотной упаковки атомов внутри белка небольшие флуктуации валентных углов (
 ) играют существенную роль в движениях, включающих другие степени свободы. При фиксации валентных углов амплитуда флуктуаций торсионных углов уменьшается в 2 раза, а конформационные переходы по торсионным углам из одного минимума энергии в другой исчезают совсем.
) играют существенную роль в движениях, включающих другие степени свободы. При фиксации валентных углов амплитуда флуктуаций торсионных углов уменьшается в 2 раза, а конформационные переходы по торсионным углам из одного минимума энергии в другой исчезают совсем.В некоторых случаях степени свободы, соответствующие изменениям значений валентных углов, учитываются неявно. Этот учет валентных углов незначительно увеличивает время счета, но значительно увеличивает конформационную подвижность, благодаря чему полученные результаты в большей степени соответствуют экспериментальным данным.
Иногда используют алгоритмы, в которых переменные, соответствующие медленным степеням свободы, постоянны на протяжении некоторого числа шагов. Однако, при таких расчетах происходит довольно быстрое накопление ошибки. Этого недостатка лишены методы MTS (multiple-time-step methods). В этих методах для вычисления сил, соответствующих быстрым и медленным степеням свободы, используются разные временные интервалы. Время счета при этом сокращается в 4 - 5 раз.
Как правило, в методе молекулярной динамике для ускорения расчетов ван-дер-ваальсовые, водородные и электростатические взаимодействия рассчитываются только между атомами, находящимися на расстоянии меньшем, чем радиус обрезания (10-15
 ). Недавно был предложен метод PPPC (particle-particle and particle-cell) расчета кулоновских взаимодействий. В методе РРРС каждый атом взаимодействует с ближними атомами путем обычных кулоновских взаимодействий, а с далеко отстоящими ячейками через общий заряд и дипольный момент этой ячейки. Размеры ячеек возрастают как функции расстояния от атома:
). Недавно был предложен метод PPPC (particle-particle and particle-cell) расчета кулоновских взаимодействий. В методе РРРС каждый атом взаимодействует с ближними атомами путем обычных кулоновских взаимодействий, а с далеко отстоящими ячейками через общий заряд и дипольный момент этой ячейки. Размеры ячеек возрастают как функции расстояния от атома: