
Опыт внедрения отечественного мдэа-абсорбента на крупнотоннажных производствах аммиака, как пример эффективной энергосберегающей технологии
| Вид материала | Документы |
- Пояснительная записка к курсовому проекту по курсу: «Процессы и аппараты химической, 61.26kb.
- Пяткова И. Н., зам директора автоматизация библиотеки вуза: опыт внедрения, проблемы,, 88.23kb.
- Эффект от внедрения системы, 46.35kb.
- Международный форум «Информационные технологии в управлении», 111.39kb.
- Программа семинара "Оптимизация и повышение эффективности работы тэс за счет внедрения, 35.35kb.
- Планирование ресурсов, включая возможность выбора статьи расходов на различные технологии., 106.76kb.
- C5- разработка энергосберегающей технологии производства магнитно-абразивных порошков, 31.2kb.
- Методы окисления в химической технологии бав, 114.42kb.
- «Харьковские стройматериалы» «фундамент» и «стены» региона, 48.33kb.
- Темы курсовых работ по дисциплине «онот», 26kb.
Механохимический синтез сложных оксидных композиций с использованием активных газовых сред
Комаров Ю.М., Смирнов Н.Н., Ильин А.П.
Ивановский государственный химико-технологический университет, Иваново, Россия,
пр. Фридриха Энгельса 7, e-mail:jek@isuct.ru
Механохимический синтез является одним из направлений нанотехнологии. Метод механохимического синтеза с использованием химически активных газовых сред это эффективный способ получения из порошков металлов нанодисперсных оксидов и сложных оксидных композиций с большой активной поверхностью, гидроксидов, индивидуальных и многокомпонентных гидроксокарбонатных солей, использующихся как предшественники ряда катализаторов. Данный метод позволяет, сократить количество стадий синтеза и избежать попадания в состав синтезируемых продуктов примесей, позволяет контролировать и регулировать состав и размер частиц получаемых продуктов, уменьшить энергетические затраты, за счет адсорбционного снижения прочности измельчаемого материала и химического взаимодействия компонентов твердой и газовой фазы в условиях механохимической активации.
В работе было изучено взаимодействие металлической меди и цинка с газовой смесью состоящей из водяного пара, аммиака и воздуха, в условиях механохимической активации в вибромельнице, при температурах 90- 100° С.
Механизм реакции газообразных компонентов с металлической медью и цинком можно представить следующим образом: газообразные компоненты, поступая в реакционный объем, адсорбируются водой, конденсируются и образуют на поверхности частиц металлов адсорбционно – конденсационный слой. При этом вначале с поверхностью металлов взаимодействует кислород, образуя металл – кислородные комплексы. В результате взаимодействия твердых компонентов с адсорбционно – конденсационным слоем содержащим аммиак идет процесс растворения металлических меди и цинка. При этом образуются неустойчивые аммиачные комплексы меди [Cu(NH3)n-xH2Ox](OH)2 и цинка, [Zn(NH3)n-xH2Ox](OH)2 при взаимодействии которых в условиях механохимической активации образуется твердый оксидный раствор. Совокупность протекающих в адсорбционно – конденсационном слое процессов можно представить следующими реакциями.
Образование аммиачного комплекса меди:
Cu + n-xNH3 + xH2O + O2 → [Cu(NH3)n-xH2Ox](OH)2
Образование аммиачного комплекса цинка:
Zn + n-xNH3 + xH2O + O2 → [Zn(NH3)n-xH2Ox](OH)2
Образование твердого раствора:
[Cu(NH3)n-xH2Ox](OH)2 + [Zn(NH3)n-xH2Ox](OH)2 → CuO·ZnO + 2n-xNH3 + 2xH2O
Оксидный твердый раствор был смешан с носителем – оксидом алюминия и после повторной механохимической обработки, полученная каталитическая композиция была испытана в реакциях низкотемпературной конверсии оксида углерода водяным паром и парового риформинга метанола. Приготовленный образец, показал достаточно высокую производительность в реакции паровой конверсии, которая достигает 11,4 мл/(г·с) при 220° С (относительно СО). Полученный катализатор проявляет высокую активностью и при получении водорода паровым риформингом метанола обеспечивая высокий выход водорода при температурах 200-250° С, при малом содержании побочного продукта – СО = 1,1 %.
РАСТВОРИМОСТЬ 1,3,5,7-ТЕТРАНИТРО-1,3,5,7-ТЕТРААЗАЦИКЛООКТАНА В ДВУХКОМПОНЕНТНЫХ РАСТВОРИТЕЛЯХ ПОЛИМЕР-НИТРОЭФИРЫ
*Коробко А.П., *Дрозд С.Н., *Крашенинников С.В., *Левакова И.В.,
**Шишов Н.И., **Бестужева Т.А.
*ФГУП «Научно-исследовательский физико-химический институт им. Л.Я. Карпова»
105064, Москва, ул. Воронцово поле, 10, E-mail: korobko@cc.nifhi.ac.ru
** Федеральный центр двойных технологий «Союз»
140090 Дзержинский Московской обл., ул. Академика Жукова, 42
1,3,5,7-Тетранитро-1,3,5,7-тетраазациклооктан (ТНТЦО) имеет ограниченную, но вполне надежно определяемую экспериментальными методами растворимость в нитроглицерине (НГ) [1]. Исследование растворимости ОК, как и других циклических нитраминов [1, 2], проводили при фиксированном содержании полиэфируретанового полимера (ПЭУ), обеспечивающем флегматизацию НГ. В то же время показано [3], что растворимость СL20 в нитраминном пластификаторе значительно уменьшается при увеличении содержания полимера в связующем.
В настоящей работе изучена зависимость растворимости ТНТЦО от концентрации ПЭУ в двухкомпонентном растворителе ПЭУ-нитроэфиры.
Объектами исследования служили частицы ТНТЦО округлой формы размером 800-900 мкм; НГ, флегматизированный разным содержанием ПЭУ (НГУ), и смесь динитродиэтиленгликоля с динитротриэтиленгликолем (СНЭ) с различным содержанием ПЭУ (СНЭУ). Методика исследования описана в [1].
Результаты кинетических измерений обрабатывали в соответствии с уравнением Шредера–Ван-Лаара
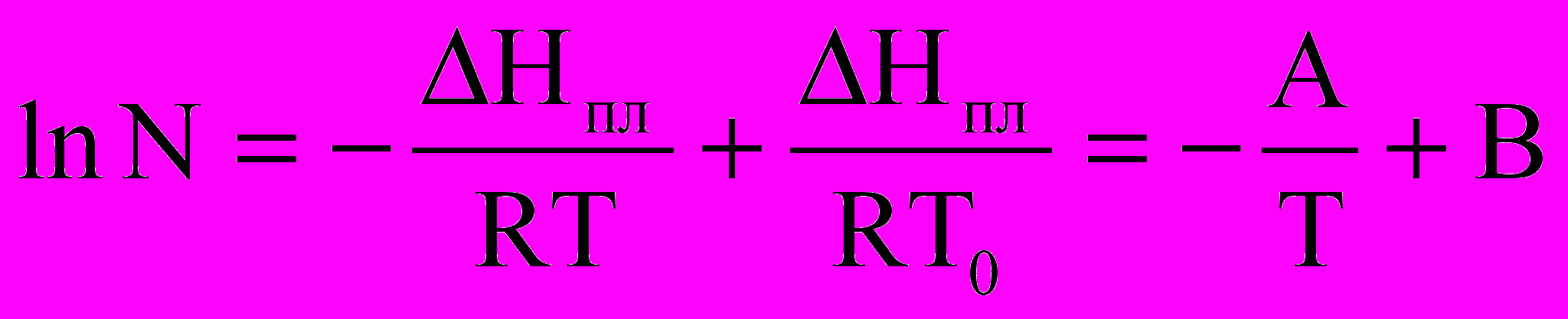 ,
,представляющим собой линейную зависимость логарифма мольной доли растворённого вещества (lnN) от обратного значения абсолютной температуры.
По экспериментальным данным вычислены значения теплот растворения ТНТЦО в исследованных системах.
Показано, что увеличение концентрации ПЭУ в двухкомпонентном растворителе приводит к заметному возрастанию растворимости ТНТЦО в СНЭУ. Растворимость же ТНТЦО в НГ при росте концентрации ПЭУ в растворителе, наоборот, уменьшается.
Теплоты растворения ТНТЦО в двух исследованных растворителях также заметно различаются.
Интерпретация полученных результатов проведена с учетом термодинамических свойств трехкомпонентных систем.
ЛИТЕРАТУРА
1. Коробко А.П., Левакова И.В., Крашенинников С.В., Дрозд С.Н. Бутенко Е.А., Бестужева Т.А., Шишов Н.И. // Вооружение. Политика. Конверсия. 2002. № 5(47). С. 69.
2. Torry S., Cunliffe A. 31th International Annual Conference of ICT (Energetic Materials), Karlsruhe, Germany, 2000, P.107.
3. Коробко А.П., Левакова И.В., Крашенинников С.В., Дрозд С.Н. Бутенко Е.А., Бестужева Т.А., Шишов Н.И. Современные проблемы пиротехники. Материалы III Всероссийской Юбилейной конференции. Сергиев Посад, 2004. С. 218.
ИССЛЕДОВАНИЕ МЕТОДОМ ДСК КИНЕТИКИ ОТВЕРЖДЕНИЯ ЭПОКСИДНЫХ КОМПОЗИЦИЙ
*Коробко А.П., *Крашенинников С.В., *Селихова В.И., **Сорина Т.Г., **Хайретдинов А.Х. **Пенская Т.В.
*ФГУП «Научно-исследовательский физико-химический институт им. Л.Я. Карпова»,
105064, Москва, Воронцово поле, 10, korobko@cc.nifhi.ac.ru
**ООО «НТИЦ АпАТэК-Дубна»,
141980, г. Дубна, МО, ул. Университетская, д. 11, стр. 16
Исследована кинетика отверждения двух составов связующего на основе эпоксидиановой смолы ЭД-20 с изо-метилтетрагидрофталевым ангидридом (МТГФА) в качестве отвердителя под влиянием ускорителей диметилдодециламина (ДМДДА) и 2-этил-4-метилимидазола (ЭМИ). Оба типа связующего используются при производстве изделий из стеклопластика пултрузионным методом в промышленных масштабах. Исследование кинетики отверждения выполнено методом дифференциальной сканирующей калориметрии (ДСК) в динамическом режиме. Основные результаты приведены в табл. 1 и 2.
Таблица 1. Кинетические параметры отверждения двух составов связующего.
| Параметр | Ускоритель | |
| ДМДДА | ЭМИ | |
| Н, Дж/г | 373.2±8.7 | 388.3±3.9 |
| п | 1.80±0.20 | 1.56±0.05 |
| ЕA, кДж/моль | 127.2±2.2 | 116.9±2.1 |
| ln Ко | 31.4±1.0 | 29.0±0.6 |
| ТП,°С | 156.3±0.7 | 148.9±0.4 |
Таблица 2. Температурные зависимости константы скорости реакции отверждения при = 0.1 и времени достижения = 0.95 для изученных составов.
| T,°C | Ускоритель | |||
| ДМДДА | ЭМИ | |||
| ln К (=0.1) | t, мин (=0.95) | ln К (=0.1) | t, мин (=0.95) | |
| 120 | -7.60 | 277.4 | -6.67 | 101.4 |
| 130 | -6.65 | 106.8 | -5.77 | 41.2 |
| 140 | -5.74 | 43.1 | -4.91 | 17.5 |
| 150 | -4.87 | 18.1 | -4.09 | 7.7 |
| 160 | -4.05 | 7.9 | -3.32 | 3.6 |
| 170 | -3.26 | 3.6 | -2.57 | 1.7 |
| 180 | -2.51 | 1.7 | -1.86 | 0.8 |
Основной вывод состоит в том, что при сопоставимых концентрациях двух изученных ускорителей ЭМИ обеспечивает системе более высокую реакционную активность и меньшую зависимость константы скорости реакции от температуры.
ВЛИЯНИЕ НЕОДНОРОДНОСТИ ПОВЕРХНОСТИ НА КИНЕТИКУ ЖИДКОФАЗНОЙ ГИДРОГЕНИЗАЦИИ МАЛЕАТА НАТРИЯ ВОДОРОДОМ, АДСОРБИРОВАННЫМ НА ПОВЕРХНОСТИ СКЕЛЕТНОГО НИКЕЛЯ
Коростелева П.О.,Улитин М.В., Лукин М.В., Филиппов Д.В.
Ивановский государственный химико-технологический университет
г. Иваново, пр. Ф. Энгельса, 7, physchem@isuct.ru
Изучение кинетики взаимодействия веществ, адсорбированных на поверхности гетерогенных катализаторов, является одним из методов определения энергетического соответствия катализатора и реагирующих веществ, и разработки методов научно-обоснованного подбора оптимальных каталитических систем. Цель настоящей работы – изучение влияния неоднородности поверхности на кинетику реакций жидкофазной каталитической гидрогенизации малеата натрия водородом, адсорбированным на поверхности скелетного никелевого катализатора.
Основным методом исследования процессов адсорбции водорода на каталитически активных поверхностях была выбрана адсорбционная калориметрия, как наиболее надежный метод определения тепловых эффектов различных физико-химических процессов. Для проведения эксперимента использовали скелетный никелевый катализатор, полученный обработкой никель-алюминиевого сплава со средним радиусом частиц 4.8 мкм 7.5 М водным раствором гидроксида натрия. Активный катализатор имел удельную поверхность 902 м2/г, пористость 0.50.05 см3/см3 и максимум функции распределения пор по радиусам, отвечающий среднему радиусу пор 20
 . Калориметрические опыты проводили в водных растворах гидроксида натрия с концентрациями 0.01, 0.1 и 1М при 303 К. В качестве гидрируемого соединения, взаимодействующего с адсорбированным водородом, использовали водные растворы малеата натрия с рН, равным водородному показателю растворителя.
. Калориметрические опыты проводили в водных растворах гидроксида натрия с концентрациями 0.01, 0.1 и 1М при 303 К. В качестве гидрируемого соединения, взаимодействующего с адсорбированным водородом, использовали водные растворы малеата натрия с рН, равным водородному показателю растворителя. Обработка полученных кинетических зависимостей с использованием уравнений формальной кинетики и моделей Рогинского-Зельдовича или Еловича, показала отсутствие явного влияния степени заполнения поверхности и концентрации гидроксида натрия на кинетические закономерности проведенных процессов, что подтверждает влияние сильной неоднородности поверхности скелетного катализатора на скорость реакции. В частности формальный порядок реакции с уменьшением степени заполнения поверхности катализатора адсорбированным водородом изменяется в пределах от 0 до 2 и не имеет четкой зависимости от рН раствора.
Однако, при обработке экспериментальных данных в линейных координатах уравнений Рогинского-Зельдовича и Еловича характер получаемых зависимостей отклонялся от линейного только при степенях заполнения близких к 0,5. Подобный вид полученных зависимостей можно объяснить следующим образом. Доказано что в водных растворах гидроксида натрия на поверхности скелетного никеля существуют как атомарные, так и молекулярные формы адсорбированного водорода. При высоких степенях заполнения скелетного никеля гидрогенизация органического реагента происходит преимущественно адсорбированным водородом с малой энергией связи с поверхностью. При низких степенях заполнения – преимущественно адсорбированным водородом с большой энергией связи с поверхностью, а при средних заполнениях (0.5) в процессе гидрогенизации участвуют все адсорбционные состояния водорода одновременно.
НАНОСВОЙСТВА ПОВЕРХНОСТЕЙ СТАЛИ И ЛЕГИРУЮЩИХ КОМПОНЕНТ ПО ИЗМЕРЕНИЯМ НА ЭСТМ
Стрючкова Ю.М., Касаткин Э.В.
ФГУП «НИФХИ им. Л.Я. Карпова», Москва, 105064, ул. Воронцово поле, 10,
e-mail - eduard.kasatkin@mail.ru
Исследованы поверхности нержавеющей стали марки Х18Н10Т и легирующих компонент (чистых хрома и никеля.)
Эксперименты проведены методами электрохимической сканирующей туннельной микроскопии (ЭСТМ) и электрохимической сканирующей туннельной спектроскопии (ЭСТС) на воздухе и в растворах 0.01 N HCl и 0.1 N H2SO4 при контролируемых потенциалах образца и иглы. Диапазон изменения потенциала для стали составлял в 0.01 N HCl от –0.46 В до +1.14 В и в 0.1 N H2SO4 от –0.06 В до +0.74 В; для хрома – в 0.01 N HCl от +0.1 В до +0.9 В и в 0.1 N H2SO4 от +0.3 В до +1.1 В; для никеля – в 0.01 N HCl от –0.01 В до +0.64 В и в 0.1 N H2SO4 от +0.184 В до +0.834 В. Все потенциалы приведены относительно н.в.э.
Получены изображения электронного профиля поверхностей Х18Н10Т, хрома и никеля с различным разрешением на воздухе и в растворах кислот.
Исследованы локальные спектральные зависимости туннельного тока от туннельного напряжения (It,Ut). И на их основе рассчитаны значения коэффициентов , β и γ, характеризующих локальные энергетические свойства поверхностей.
Обнаружено, что потенциал образца оказывает более заметное влияние на электрофизические свойства, определяющие туннельный перенос электронов, чем на нанорельеф.
В целом поверхность в растворе при заданном потенциале выглядит более энергетически однородной, чем на воздухе, где наблюдается большая дисперсия свойств.
Для Х18Н10Т при измерениях в растворах, как и при измерениях на воздухе, обнаружено, что энергетические свойства отдельных мест поверхности на атомном уровне определяются взаимным влиянием ближайших атомов, входящих в ее состав, – наличием матричного эффекта.
Исследования показали, что локальные наносвойства поверхности обладают большой изменчивостью во времени, т.е. отдельные места поверхности существенно нестабильны, что проливает новый свет на природу коррозионной устойчивости материалов.
Работа выполнена при поддержке РФФИ, проекты №№ 04-03-32337а и 08-03 00453а
СТАДИИ ГОМОГЕННЫХ ПРЕВРАЩЕНИЙ
В УСЛОВИЯХ ЖИДКОФАЗНОЙ ГЕТЕРОГЕННО–КАТАЛИТИЧЕСКОЙ ГИДРОГЕНИЗАЦИИ
Немцева М.П., Тюрина Ю.М., Лефëдова О.В.
Ивановский государственный химико-технологический университет
г. Иваново, пр. Ф. Энгельса, 7, Email: physchem@isuct.ru
Известно, что подавляющее число реакций жидкофазной гидрогенизации органических соединений представляют собой многостадийные процессы, селективность которых по отношению к целевому продукту определяется соотношением скоростей отдельных стадий.
Особое место среди многообразия гетерогенно-каталитических процессов занимают реакции гидрогенизации замещенных 2'-гидроксинитроазобензолов – исходных соединений синтеза эффективных фотостабилизаторов полимеров. Наличие нитро- и азогрупп в молекулах гидрируемых соединений приводит к увеличению числа стадий на поверхности катализатора, а их положение относительно друг друга способствует параллельному протеканию гомогенных перегруппировок промежуточных продуктов гидрогенизации названных групп в объеме раствора. Последнее обстоятельство является отличительной чертой указанного процесса, так как, согласно данным литературы, механизмы превращения исходных соединений в реакциях восстановительного и окислительного катализа включают исключительно гетерогенные стадии. Одновременное протекание гетерогенных и гомогенных стадий при гидрогенизации замещенных 2'-гидроксинитроазобензолов предполагает возможность регулирования селективности реакции по отношению к целевым продуктам – производным 2'-гидроксибензотриазолов и их N-оксидам – варьированием природы и состава растворителя.
Кинетические исследования проводили в реакторе с интенсивным перемешиванием жидкой фазы, исключающим влияние внешнего массопереноса на скорость гидрогенизации. В качестве катализатора использовали доступный и дешевый скелетный никелевый катализатор (никель Ренея). Аналитический контроль за ходом гидрогенизации осуществляли посредством комплексного метода, основанного на сочетании тонкослойной хроматографии и спектрофотометрии, который позволяет фиксировать концентрации продуктов реакции, претерпевающих гомогенные превращения. Продукты реакции – производные 2'-гидроксибензотриазолов и их N-оксиды были получены в чистом виде. У синтезированных образцов были определены физико-химические константы, которые соответствовали характеристикам, приведенным в литературе.
Экспериментально доказано, что определяющим фактором в изменении скоростей стадий гомогенных превращений промежуточных продуктов гидрогенизации замещенных 2-гидроксинитроазобензолов является введение в индивидуальные или водные растворы органических растворителей кислотных или основных добавок. В частности, в присутствии гидроксида натрия скорость бензотриазольной циклизации замещенных 2'-гидроксинитрогидразобензолов с образованием соответствующих N-оксидов в объеме раствора увеличивается более чем на два порядка и приближается к значениям скоростей гетерогенных стадий. Таким образом, существенное изменение соотношения скоростей гетерогенных и гомогенных стадий реакции можно достичь изменением жидкофазной составляющей каталитической системы при использовании одного и того же гетерогенного катализатора. Варьирование состава растворителя посредством введения небольших количеств добавок позволяет изменять селективность каталитической гидрогенизации замещенных 2'-гидроксинитроазобензолов по отношению к целевым продуктам в пределах от 25 до 85 %.
Калориметрическое и хемосорбционное исследование хемосорбции Н2, О2, СО2 и Н2О на Cr2O3 и влияние хемосорбции Н2 на каталитическую активность Cr2O3 в реакции дегидрирования пропана
В.Е. Островский, Е.А. Кадышевич, Б.В. Гостев, Ю.А.Агафонов, А.Л. Лапидус
Оксид хрома широко используют как однокомпонентный катализатор и как компонент Cr2O3/Al2O3 или более сложных катализаторов, ускоряющих различные окислительно-восстановительные реакции такие как дегидрирование С3Н8 и С2Н4 в присутствии СО2, окислительное дегидрирование изобутана, синтез метанола и шифт-реакцию и др.
В реакциях, протекающих в присутствии катализаторов, содержащих оксид хрома, компонентами реагирующих газовых смесей зачастую являются Н2, Н2О и СО2; в ходе каталитических процессов, протекающих в восстановительной среде, поверхность оксида хрома может быть в большей или в меньшей степени окислена.
Поэтому для понимания механизма каталитических реакций, протекающих в присутствии оксида хрома, важно знать механизм хемосорбции, соотношение между хемосорбционными и сорбционными процессами при различных температурах, влияние хемосорбции каждого из перечисленных веществ на хемосорбцию каждого другого из них. Хотя механизм хемосорбции газов и паров на оксиде хрома исследовали различные авторы, в том числе - в нашей стране, многие вопросы остаются невыясненными, а некоторые – не изучались вовсе в связи с методическими трудностями. Например, в литературе нет количественных данных о влиянии степени окисленности поверхностного слоя на параметры хемосорбции газов и паров и о поглощении компонентов реакционных газовых смесей в объем оксида хрома; мольные теплоты хемосорбции газов и паров на Cr2O3 измеряли лишь70 лет назад при комнатной температуре на методическом уровне, не отвечающем современным требованиям.
В данной работе приведены оригинальные данные о мольных тепловых эффектах и о кинетике хемосорбции и сорбции перечисленных выше газов и паров, полученные в широких интервалах изменения условий экспериментов. Опыты по хемосорбции и сорбции выполнены при 300–800 К, а калориметрические опыты – при 300–570 К. Впервые каждый эксперимент характеризуется не только температурой и количеством изучаемого газа в катализаторе, но также – содержанием в нем других хемосорбированных и сорбированных газов. Наряду с хемосорбционными данными, работа содержит результаты экспериментов по дегидрированию С3Н8 до С3Н6 в различных стационарных условиях в присутствии Cr2O3.
Совокупность полученных экспериментальных результатов обсуждается в контексте проблем механизма и оптимизации условий дегидрирования и гидрирования углеводородов в присутствии Cr2O3 и в контексте фундаментальных проблем гетерогенного катализа.
ТЕХНОЛОГИЯ КАТАЛИТИЧЕСКОЙ ПЕРЕРАБОТКИ КУБОВЫХ ОТХОДОВ, СОДЕРЖАЩИХ ЧЕТЫРЕХХЛОРИСТЫЙ УГЛЕРОД, В ХЛОРИСТЫЙ МЕТИЛ
Перишкова Е.В., Занавескин Л.Н.
Федеральное государственное унитарное предприятие Научно-исследовательский физико-химический институт имени Л.Я. Карпова, г. Москва
г. Москва, ул. Воронцово поле, 10., e-mail: epershikova@mail.ru
Четыреххлористый углерод (ЧХУ) до недавнего времени являлся одним из важнейших продуктов промышленного хлорорганического синтеза. Но в соответствии с Программой Организации Объединенных Наций по окружающей среде ЧХУ вместе с рядом других хлорорганических соединений отнесен к озоноразрушающим веществам, и его выпуск как товарного продукта в настоящее время прекращен.
Одним из способов переработки ЧХУ и кубовых отходов, содержащих ЧХУ, является процесс его каталитического взаимодействия с метанолом, в результате которого образуется хлористый метил.
В качестве катализаторов процесса могут быть использованы галогениды и оксиды меди, магния, кальция, бария, цинка, кадмия, ртути, хрома, молибдена, марганца, железа, кобальта и никеля, нанесенные на активированный уголь или другой пористый носитель. Предпочтение отдается хлористому цинку при его содержании на активированном угле или оксиде алюминия 20-25% масс.
С целью исследования стабильности работы катализаторов, приготовленных на основе активированного угля и у- оксида алюминия в процессе взаимодействия ЧХУ с метанолом были проведены серии экспериментов с использованием в качестве сырья как чистого четыреххлористого углерода, так и осветленных кубовых продуктов действующего производства хлорметанов.
Катализатор на активированном угле при работе на осветленных кубовых продуктах показал практически ту же конверсию реагентов, что и при работе на чистом ЧХУ. При этом следует отметить, что конверсия хлорэтанов и хлорэтиленов, содержащихся в кубовых продуктах, была не полной, и они всегда присутствовали в продуктах реакции.
Катализатор на основе γ-оксида алюминия на чистом ЧХУ проработал с постоянной активностью 180 часов, затем наблюдалось резкое снижение активности. При работе на осветленных кубовых продуктах активность этого катализатора монотонно снижалась практически с первых часов работы. Через 250 часов конверсия ЧХУ уменьшилась с 98 до 42%.
Таким образом, наилучшую стабильность показал катализатор на основе активированного угля, что явилось основанием рекомендовать его для проведения всех дальнейших исследований процесса переработки ЧХУ в хлористый метил.
На основе проведенных экспериментальных исследований определены оптимальные условия проведения процесса переработки четыреххлористого углерода и содержащих его отходов на катализаторе 25% хлористого цинка, нанесенного на активированный уголь: температура 180-210°С, температура горячей точки до 230 С, время контакта 20-25 секунд, мольное соотношение ЧХУ:метанол 1:4,1-4,2.
По результатам выполненных экспериментальных работ и расчетов с учетом скорости снижения активности катализатора был определен необходимый его объем для загрузки реактора переработки осветленных кубовых отходов производства хлорметанов, разработана технологическая схема процесса.
Применение продетонаторов для активации углеводородов
к детонационному горению
Петрухин Н.В., Загарских В.И., Волков А.В.
Военная академия имени Петра Великого,
г. Москва 109074, Китайгородский проезд, 9/5
E-mail: vladmelva@rambler.ru
Исследовано действие активирующих добавок – продетонаторов – на условия и критические параметры инициирования углеводородных топлив. На основе анализа теоретического материала, имеющегося в литературе, показано, что в качестве активирующих присадок могут быть использованы пероксиды, нитраты и альдегиды. Разработана и изготовлена экспериментальная установка для определения критических параметров инициирования. Экспериментально установлено, что наибольшую эффективность проявляют пероксиды водорода и бензола. Если при ударно-волновом нагружении энергия инициирования в парах керосино- и н-гептанововоздушных смесей при 250°С составляет (1,56…3,60)·106 Дж/м2, то при энергии внешнего воздействия в 50 Дж добавление пероксида водорода обеспечивает детонацию топливно-воздушной смеси (ТВС) даже на основе дизельного топлива. Одновременно показано, что использование продетонаторов существенно уменьшает предетонационный участок в углеводородных топливных смесях.
Для проведения исследования был создан испытательный стенд. Стенд состоит из трех основных систем – силовой, контрольно-измерительной и рабочей камеры, моделирующей камеру сгорания ИДД.
Была исследована активирующая способность веществ. В качестве ТВС использовали стехиометрическую смесь паров дизельного топлива ДТ-3-воздух. Все выбранные соединения достаточно легко диссоциируют на свободные радикалы:
| ПБ 2C6H5COO; ПА ® 2(СH3)2COO; | ИН ® (CH3)2CHO + NO2; ИБН ® 2(CH3)2CCN. |
Активирующий эффект выбранных соединений обусловлен заменой первичного разложения углеводорода горючего более энергетически выгодной реакцией разложения продетонатора. Разложение продетонаторов происходит с энергией активации 0,15…0,19 МДж/моль вместо 0,37…0,49 МДж/моль в случае углеводородов. Кроме того, наряду с первичным разложением углеводородов возможна замена углеводородных радикалов атомами кислорода, которые служат центрами высокотемпературного воспламенения.
Относительная активность выбранных соединений была оценена по периоду задержки воспламенения delay при слабом инициировании искрой.
Полученные в работе уровни delay при введении продетонаторов в 2-3 раза ниже delay исходного горючего. Например delay для цетана при 300 С составляет около 40 мс, для дизельных фракций при 440 °С tdelay находится в интервале 10…40 мс. С увеличением температуры tdelay резко сокращается.
В целом следует отметить, что введение в ТВС продетонаторов активно влияет на скорость взрывчатого превращения. При отработке рецептуры ТВС, включающей продетонатор и одновременно удовлетворяющей своими эксплуатационными показателями, можно получить топливо с приемлемой детонационной активностью для импульсного детонационного двигателя.
РАЗМЕРНЫЙ ЭФФЕКТ В СЛОЖНЫХ УЗКОПОРИСТЫХ СИСТЕМАХ
И ПРОБЛЕМЫ АДСОРБЦИОННОЙ ПОРОМЕТРИИ
А.Г. Петухов, Ю.К. Товбин
ГНЦ РФ “НИФХИ им.Л.Я.Карпова”, Москва, Воронцово Поле 10, tovbin@cc.nifhi.ac.ru
Основным методом определения пористости дисперсных материалов является метод адсорбционной порометрии, в котором экспериментальные данные по изотермам адсорбции и их гистерезисным петлям интерпретируются с помощью уравнения Кельвина, которое связывает величины давлений насыщенных паров в пористых телах с характерным размером пор и поверхностным натяжением на границе раздела жидкость - пар адсорбируемых молекул.
Исследования фазовых диаграмм адсорбатов в мезопористых системах методами статистической термодинамики подтвердили экспериментальный факт, что по мере уменьшении диаметра цилиндрических пор пропадет петля на изотермах адсорбции. Теоретическое объяснение этого факта получено только в рамках модели решеточного газа с использованием калибровочных функций, которые повышают точность расчетов как вблизи критической температуры, так и при температурах ниже критической, когда в узких порах учитывается влияние размерного фактора. В цилиндрических порах размерный вклад в калибровочную функцию был рассчитан на основе концепции квазиодномерности, что позволило описать экспериментальные данные адсорбции в мезопористых адсорбентах МСМ-41 для Ar и N2.
В данной работе разработаны теоретические основы учета размерного фактора в более сложных узкопористых системах: сфероцилиндрических, глобулярных и щелевидных. Использовано два типа условий для формулировки условий, в которые невозможны фазовые переходы первого рода. Критерий первого типа построен по аналогии с критерием квазиодномерности в цилиндрических порах через отношение вкладов центральной и поверхностных областей поры. Второй тип критерия использует найденные условия для реализации фазовых переходов в бесконечно длинных цилиндрических порах для ограничения протяженности участков поры, аппроксимирующих длинные каналы. Оба типа критерия зависят от характера протяженности потенциала адсорбент - адсорбат. Разработан подход для связи величины размера локального участка поры с параметрами размерной части калибровочной функции.
Обсуждается сравнение результатов для функции распределения пор по размерам, полученных с помощью уравнения Кельвина и молекулярно-статистической теории на основе модели решеточного газа. Показано, что теория объясняет отсутствие гистерезисных петель с уменьшением размера пор и дает правильный критический размер, ниже которого отсутствует капиллярная конденсация. Сформулированы молекулярные подходы к разработке метода определения распределения пор по размерам по данным адсорбционной порометрии для мембран, представляющих собой сложных пористых систем, состоящих из пор разного размера и разной геометрии.
Определена нижняя граница характерных размеров цилиндрических пор, для которых применимо уравнение Кельвина. Результаты расчетов сопоставлены с экспериментальными данными и с результатами численных исследований методами Монте-Карло и молекулярной динамики.
Работа выполнена при поддержке РФФИ (код проекта № 06-03-32031а ).
ВЛИЯНИЕ РАЗМЕРА И СТРОЕНИЯ КЛАСТЕРОВ ЗОЛОТА
НА ИХ КАТАЛИТИЧЕСКУЮ АКТИВНОСТЬ
Пичугина Д.А., Ланин С.Н., Викулина А.С., Петров Н.Ю.,
Шестаков А.Ф., Кузьменко Н.Е.
Химический факультет МГУ имени М.В. Ломоносова, Москва, Ленинские горы, д. 1, стр. 3, 119991, daria@phys.chem.msu.ru
Наночастицы золота нашли широкое применение в катализе как эффективные катализаторы низкотемпературного окисления и гидрирования углеводородов [1]. Природа активных центров Aun до сих пор точно не установлена. Не разработана и теория, объясняющая взаимное влияние металлов в смешанных наноразмерных системах, приводящее к резкому увеличению их активности. В работе квантово-химическим методом изучено строение смешанных катализаторов на примере простейших систем NiAun и PdAun (n=1–4), рассмотрено взаимодействие углеводородов с кластерами золота различного строения. Расчет проводился методом DFT PBE, использовался расширенный базисный набор с псевдопотенциалом SBK [2].
По данным расчета замена атома золота в кластерах Aun на никель или палладий приводит к уменьшению расстояний между атомами в кластере в среднем на 0.2 Å без существенного изменения морфологии частиц. Для биметаллических NiAun и PdAun, так же как и для Aun, характерна плоская структура. В замещенных кластерах наблюдается уменьшение длины активного периметра, следовательно, можно ожидать увеличение их реакционной способности [3]. Для проверки полученного вывода был проведен расчет структуры комплексов СmH2m+2–NiAun. Из таблицы видно, что присутствие никеля в кластере увеличивает адсорбционную способность кластеров золота более чем в два раза.
Рассчитанная энергия связи алкан–Aun и алкан–NiAun в кДж/моль
| Углеводород | Au3 | Au4 | Au5 | Au2Ni | Au3Ni | Au4Ni |
| CH4 | –27.2 | –19.9 | –1.5 | –77.4 | –52.6 | –51.5 |
| C2H6 | –31.5 | –25.9 | –12.1 | –107.6 | –49.5 | –54.6 |
| C3H8 | –34.9 | –29.2 | –16.1 | –97.5 | –62.4 | –55.0 |
Моделирование активации алканов на тетраэдрическом кластере Au20 показало, что повышенной адсорбционной и каталитической активностью обладают координационно-ненасыщенные атомы в вершине частицы. При увеличении количества атомов в углеродной цепи наблюдается рост (по модулю) энергии связи Au20–CnH2n+2 от –3.1 кДж/моль для метана до –19.9 кДж/моль для октана. На адсорбционные способности кластера влияет и его дефектность: так энергия связи пентана с Au22 составила –22.5 кДж/моль, тогда как с Au20 –16 кДж/моль.
Проведенное исследование показало, что углеводороды активируются преимущественно на вершинных атомах кластера, управление этим процессом возможно путем введения постороннего металла и изменением структуры кластера золота.
Работа поддержана грантом Президента РФ для поддержки кандидатов наук и их научных руководителей (МК-3156.2007.3) и грантом РФФИ (№ 06-03-33131-а).
1. A.S.K. Hashmi, G.J. Hutchings, Angew. Chem. Int. Ed. Engl., 2006, 45, 7896.
2. D.A. Pichugina, A.F. Shestakov, N.E. Kuz’menko, Gold Bull., 2007, 40, 115.
3. S.N. Rashkeev, A.R. Lupini, S.H. Overbury, Phys. Rev. B, 2007, 76, 035438.
ИССЛЕДОВАНИЕ ЖИДКОФАЗНОЙ ГИДРОГЕНИЗАЦИИ МАЛЕАТА НАТРИЯ НА ЧАСТИЧНО ДЕЗАКТИВАРОВАННОМ СКЕЛЕТНОМ НИКЕЛЕ
Прозоров Д. А., Лукин М. В., Улитин М. В.
Ивановский государственный химико-технологический университет
г. Иваново, пр. Ф. Энгельса, 7, physchem@isuct.ru
Известно, что основной причиной, влияющей на активность гетерогенных катализаторов и свойства каталитических систем на их основе, является изменение адсорбционных состояний веществ-участников реакции. Поверхностные концентрации адсорбированных состояний водорода определяют скорость и селективность реакций жидкофазной гидрогенизации. Величины адсорбции индивидуальных форм водорода зависят от природы катализатора, природы и состава растворителя и могут целенаправленно изменяться введением в реакционную систему каталитических ядов.
Цель настоящей работы – исследование процесса жидкофазной гидрогенизации малеата натрия на скелетном никеле, поверхность которого была частично дезактивирована сульфидом натрия.
Для проведения эксперимента использовали скелетный никелевый катализатор, полученный обработкой никель-алюминиевого сплава со средним радиусом частиц 4.8 мкм 7.5 М водным раствором гидроксида натрия. Активный катализатор имел удельную поверхность 902 м2/г, пористость 0.50.05 см3/см3. В качестве гидрируемого соединения, использовали малеат натрия. Все опыты проводили в водных растворах гидроксида натрия с концентрациями 0.01, 0.1 и 1М при 303 К. В качестве реагента использовали водные растворы малеата натрия с рН, равным водородному показателю растворителя. Частичную дезактивацию катализатора проводили обработкой последнего различными количествами 0,5 М водного раствора сульфида натрия. Предварительные опыты показали, что при низких концентрациях сульфид-ионы необратимо взаимодействуют с активными центрами поверхности таким образом, что остаточные количества ионов S2- не могут быть определены аналитически.
В результате проведенных экспериментов были получены кинетические кривые зависимости скорости реакции от количества поглощенного водорода, а
 также теплоты гидрогенизации малеата натрия водородом, адсорбированным на поверхности частично дезактивированного пористого никелевого катализатора.
также теплоты гидрогенизации малеата натрия водородом, адсорбированным на поверхности частично дезактивированного пористого никелевого катализатора. Обработка полученных кинетических зависимостей в линейных координатах классического метода Вант-Гоффа, свидетельствует о сложности кинетического механизма, протекающих процессов. По всей видимости увеличение содержания сульфид-ионов приводит к частичному заполнению вакантных d-орбиталей никеля неподеленными электронными парами сульфид-ионов и заметному снижению порядка реакции и наблюдаемой константы скорости.
С другой стороны увеличение исходной концентрации сульфида натрия в растворе первоначально приводит к уменьшению теплот гидрогенизации малеата натрия, что объясняется взаимодействием ионов S2- с активными центрами поверхности, обладающими наименьшей энергией связи с адсорбированным водородом.
Таким образом, совместный анализ кинетических и калориметрических данных показал, что введение сульфида натрия в объемную фазу реакций жидкофазной гидрогенизации способно существенным образом изменять поверхность скелетного никеля, тем самым, меняя адсорбционные состояния водорода как основного вещества-участника реакции жидкофазной гидрогенизации.
Механохимический Синтез алюмокалиевого
носителя катализатора конверсии метана
Прокофьев В.Ю., Кузнецов В.В.
Ивановский государственный химико-технологический университет, г. Иваново
153000, г. Иваново, пр. Ф.Энгельса, 7. E-mail: pv@isuct.ru
Для проведения высокотемпературных процессов используют никелевые катализаторы, носителем которых является оксид алюминия. Для увеличения селективности в состав катализатора вводят щелочные металлы. Повышение механической прочности гранул добиваются добавлением высокоглиноземистого цемента. Весьма интересной представляется перспектива создания носителя катализатора, где на стадии приготовления синтезируется алюминат калия. В качестве сырья были использованы глинозем и гидраргиллит. Источником калия служил твердый КОН. Активирование твердых смесей проводили в вибрационной ролико-кольцевой мельнице с ударно-сдвиговым характером нагружения.
Непосредственно после совместного механоактивирования глинозема и гидроксида калия, содержание которого составляло от 2 до 5 мас.%, образование новых фаз не обнаружено. Добавка 2 мас.% КОН в твердом виде на стадии измельчения с последующим прокаливанием приводит к появлению на рентгенограммах серии рефлексов, отличных от -Al2O3. По набору рефлексов и соотношению интенсивностей характеристических пиков можно сделать вывод, что образец кроме -Al2O3 содержит такие алюминаты калия, как K2O·9Al2O3 (K0.67Al6O9.33) и K2O·11Al2O3 (KAl11O17). Здесь и далее порядок перечисления фаз соответствует уменьшению их содержания. К сожалению, все наиболее интенсивные рефлексы располагаются очень близко друг к другу, и поэтому не представляется возможным определить их точное содержание в образце. При увеличении калийной составляющей до 5 мас.% КОН на дифрактограммах наблюдается появление других рефлексов в тех же областях углов дифракции. Это можно связать с образованием в системе помимо K2O·9Al2O3 и K2O·11Al2O3 алюмината состава 3K2O·16Al2O3 (K2Al10,67O17). В случае использования вместо твердого КОН его раствора качественных изменений в рентгеновском спектре не обнаружено, наблюдается лишь перераспределение интенсивностей. Это говорит о том, что среди алюминатов преобладающей фазой является K2O·11Al2O3, остальные же упомянутые выше соединения калия образуются в меньшем количестве. Для образца, в котором Al2O3 введен в форме гидроксида, на дифрактограмме присутствуют достаточно интенсивные рефлексы K2O·11Al2O3. Синтез других алюминатов в этом случае весьма незначителен. Необходимо особо отметить важную деталь, характерную для всех образцов. Согласно данным ASTM, у алюминатов калия должны наблюдаться очень интенсивные пики, отвечающие межплоскостным расстояниям более 1,1 нм. На экспериментальных же рентгенограммах в этой области наблюдаются весьма слабые рефлексы. Другой особенностью является значительное уширение всех пиков. Вкупе эта картина может быть интерпретирована как образование алюминатов калия с плохо окристаллизованной структурой, которая имеет лишь ближний порядок.
На прокаленные носители методом пропитки был нанесен никель в оксидной форме. Готовый катализатор после восстановления в токе водорода был испытан в реакции паровой конверсии метана (соотношение пар:газ = 2:1) при объемной скорости 6000 ч–1. Максимальную активность, в том числе и при низких температурах, показал катализатор, носитель которого был приготовлен с использованием совместного активирования глинозема и КОН.
ВЛИЯНИЕ СВОЙСТВ ПОВЕРХНОСТИ АДСОРБЕНТОВ
НА ОЧИСТКУ ЭКСТРАКЦИОННОЙ ФОСФОРНОЙ КИСЛОТЫ
И.Г. Пухов, Н.Н.Смирнов, Н.Е. Гордина
Ивановский государственный химико-технологический университет
153000 г. Иваново, пр. Ф. Энгельса 7, каф. ТНВ. Е-mail: Smirnov@isuct.ru
Наиболее эффективным способом очистки ЭФК является удаление соединений фтора (HF, SiF4) в газовую фазу при упаривании с продувкой газового теплоносителя. Применение адсорбента позволяет увеличить скорость удаления фтористых соединений в газовую фазу и повысить эффективность осаждения соединений Si, Fe, Al, Ca, Mg в твердую.
Адсорбционную очистку ЭФК проводили в лабораторных условиях при соотношении уголь/кислота = 1 : 100 (t=80-100 °С; =2,0-2,5 часа), совмещая процессы упаривания (концентрирования) ЭФК и отдувки фтористых соединений горячим воздухом при интенсивном перемешивании в реакторе, в который одновременно помещен адсорбент. В качестве адсорбентов использовали активированный уголь марки БАУ, природный графит и сажа.
Указанные адсорбенты значительно различаются размером удельной поверхности. Угли марки БАУ обладают удельной поверхностью до 1000 г/м2, в то время как графит и сажа имеют удельную поверхность, не превышающую 100 г/м2. Тем не менее, степень очистки, в частности, от соединений фтора, на указанных адсорбентах идентична. Они позволяют при указанных условиях опыта получить экстракционную фосфорную кислоту марки Т2. В связи с этим, можно сделать вывод, что в адсорбции примесей из раствора ЭФК микропоры задействованы незначительно из-за больших размеров адсорбируемых молекул. Ведущую роль в процессе адсорбционной очистки ЭФК играют мезопоры, то есть фактически, геометрическая поверхность адсорбента. При этом происходит хемосорбция примесей за счет кислотно-основного взаимодействия с активными центрами на поверхности угля.
Установлено, что такими центрами являются поверхностные оксиды. Присутствие на поверхности исследуемых углеродных материалов кислородосодержащих функциональных групп подтверждается данными ИК-спектроскопии образцов. Обнаружены карбоксильные, карбонильные, хиноновые, гидроксильные группы. При этом группы основного характера эффективны при сорбции соединений кремния, а кислотные – фтора, алюминия, железа. На степень очистки кислоты будет влиять концентрация указанных кислотно-основных центров.
Таким образом, можно отметить, что основным способом увеличения адсорбционной способности адсорбента по отношению к примесям в растворе ЭФК является не простое увеличение его удельной поверхности, а модифицирование поверхности, направленное на построение на углеродной матрице развитой системы поверхностных оксидов, отвечающих за процесс адсорбции соединений фтора, алюминия, железа, кремния.