
А. С. Пушкина Исследовательская работа Исследование
| Вид материала | Исследовательская работа |
- Тема: «Любви все возрасты покорны» Любовная лирика А. С. Пушкина. Урок- открытие Цели, 56.7kb.
- Исследовательская работа по теме: Молодежная политика в Республике Беларусь на современном, 222.46kb.
- А. С. Пушкина На конференцию «Первые шаги в науку» Научно-исследовательская работа, 669.28kb.
- Г. Александрова Исследовательская работа по творчеству А. С. Пушкина, 146.64kb.
- Исследовательская работа по литературе автор: ученица 7 класса, 92.87kb.
- Муниципальное общеобразовательное учреждение, 231.01kb.
- А. С. Пушкина Метафорические розы А. С. Пушкина Исследовательская работа, 92.38kb.
- Томска Город Томск, ул. Бирюкова, 22 (8-382) 67-88-78 Роль фразеологических оборотов, 172.33kb.
- Т. И. Алиев Учебно-исследовательская работа И1 Исследование, 295.14kb.
- Исследовательская работа Исследовательская работа, 254.75kb.
Конференция одаренных школьников Intel-Авангард 2006
Брянский городской лицей № 1 имени А. С. Пушкина
Исследовательская работа
Исследование эффективности применения сильных
и слабых оснований для определения концентрации
дикарбоновых кислот кондуктометрическим методом
Выполнил: ученик 10 медико-биологического класса Мадуар Салим
Научные руководители: кандидат химических наук, преподаватель Брянского
государственного университета
Гегеле Ф. Ф.,
учитель химии Брянского городского
лицея №1 Геращенков А. М.
Брянск 2006
Содержание
Ведение…………………………………………………………………………………….……3
1.Литературный обзор…………………………………………………………………………4
1.1.Электропроводность растворов электролитов…………………………………………...4
1.2.Теория Дебая-Онзагера…………………………………………………………………….5
1.3. Аномальная подвижность ионов гидроксония и гидроксила…………………………...7
1.4. Определение степени и константы диссоциации слабого электролита
по электрической проводимости раствора................................................................................8
1.5. Определение эквивалентной электропроводности слабого электролита при
бесконечном разведении. Расчет константы диссоциации по методу Фуосса и Брея……..8
.2.Экспериментальная часть………………………………………………………...………….9
2.1.Методика измерения электропроводности………………………………………………..9
2.2.Кондуктометрическое титрование…………………………….………………………….10
3.Обсуждение результатов……………………………………………………………………12
3.1.Титрование щавелевой кислоты……………………………………………………….….12
3.2.Титрование янтарной кислоты……………………………………………………………14
3.3.Титрование яблочной кислоты…………………………………………………………...15
Выводы…………………………………………………………………………………………21
Литература……………………………………………………………………………………..22
Введение
Карбоновые кислоты представляют особый интерес среди органических соединений: они широко распространены в растительных и животных объектах, разнообразны по составу и химическому строению, и, пожалуй, самое главное, принимают активное и непосредственное участие в обмене веществ. Они являются промежуточными звеньями распада углеводов при дыхании и необходимы для синтеза аминокислот. Особенно следует выделить ди- и трикарбоновые кислоты, такие как щавелевая, янтарная, яблочная, винная, лимонная. Общая кислотность одна из важнейших биохимических характеристик растений, она является сортовым и видовым признаком, а также может служить показателем степени созревания плодов [2].
Существуют различные методы определения содержания кислот в водных растворах, но наиболее распространены титриметрические методы анализа [6]. Они достаточно просты в применении. Однако визуальное фиксирование конечной точки титрования по изменению окраски индикатора не всегда позволяет говорить о высокой степени надежности полученных результатов. При этом конечная точка титрования чаще всего не совпадает с точкой эквивалентности и определяется своим интервалом рН-перехода окраски конкретного индикатора [7]. При титровании очень разбавленных растворов вероятность ошибки еще больше увеличивается, потому что необходимо учитывать ту часть кислоты, которая непосредственно реагирует с основными центрами самого индикатора.
В этом смысле кондуктометрическое титрование является гораздо более эффективным и надежным методом [8]. Он основан на измерении удельной электропроводности титруемого раствора, которая изменяется в процессе титрования в результате протекающих в системе химических реакций. Построение кривой титрования позволяет определить точку эквивалентности по характерному излому. К преимуществам данного метода следует отнести его высокую чувствительность, которая позволяет анализировать очень разбавленные растворы как сильных, так и слабых органических и неорганических кислот. Особо следует подчеркнуть, что данный метод позволяет анализировать мутные и окрашенные растворы, что часто невозможно при титровании с традиционными индикаторами. Следует заметить, что кондуктометрия одноосновных кислот хорошо разработана и изучена. В то же время в литературе практически не встречаются работы, посвященные кондуктометрическому анализу дикарбоновых кислот.
Целью настоящей работы явилось исследование возможности применения кондуктометрического титрования для определения содержания дикарбоновых кислот в водных растворах.
1. Литературный обзор
1.1. Электропроводность растворов электролитов
Р
 астворы солей, кислот и оснований обладают способностью проводить электрический ток. Это является следствием электролитической диссоциации, то есть распад нейтральных молекул на положительно заряженные ионы (катионы) и на отрицательно заряженные ионы (анионы).Скорость иона в растворе при E=1в/см называют абсолютной подвижностью иона.
астворы солей, кислот и оснований обладают способностью проводить электрический ток. Это является следствием электролитической диссоциации, то есть распад нейтральных молекул на положительно заряженные ионы (катионы) и на отрицательно заряженные ионы (анионы).Скорость иона в растворе при E=1в/см называют абсолютной подвижностью иона.Подобно проводникам первого рода растворы электролитов подчиняются закону Ома.
I = E/ = E. []= Ом-1см-1 [1.1]
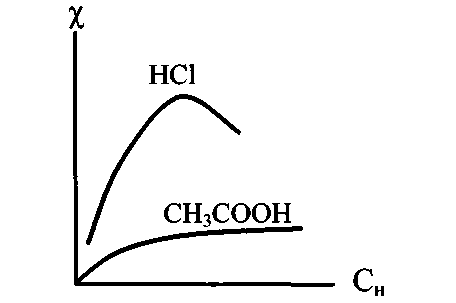
– удельная электропроводность. Эта величина обратно пропорциональна сопротивлению столба раствора длиной 1см. и площадью поперечного сечения 1 см2. Удельная электропроводность зависит от индивидуальных свойств ионов и концентрации раствора (рис.1-1).
Рис. 1-1. Зависимость удельной
электропроводности растворов
электролитов от концентрации
В разбавленных растворах сильных электролитов рост электропроводности обусловлен увеличением концентрации ионов. Однако в концентрированных растворах с ростом концентрации увеличиваются силы электростатического взаимодействия между ионами, и электропроводность уменьшается.
П
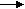 ри определенном значении концентрации влияние уменьшения скорости больше влияния увеличения концентрации ионов. В результате электропроводность уменьшается. В растворах слабых электролитов силы электростатического притяжения малы. Скорость движения ионов не зависит от концентрации. В разбавленных растворах, где → 1, небольшой рост удельной электропроводности с увеличением концентрации объясняется увеличением концентрации ионов.
ри определенном значении концентрации влияние уменьшения скорости больше влияния увеличения концентрации ионов. В результате электропроводность уменьшается. В растворах слабых электролитов силы электростатического притяжения малы. Скорость движения ионов не зависит от концентрации. В разбавленных растворах, где → 1, небольшой рост удельной электропроводности с увеличением концентрации объясняется увеличением концентрации ионов.С

 умма ионов обоих знаков в 1 см3 электролита диссоциирующего так:
умма ионов обоих знаков в 1 см3 электролита диссоциирующего так:АB → A+ + B- - выражается формулой:
n+ + n- = 2CN/1000 [1.2]
где NA – число Авогадро, С- концентрация растворённого вещества и α- степень диссоциации.
При  1 С0, а при возрастании концентрации 0. В обоих случаях удельная электропроводность слишком мала.
1 С0, а при возрастании концентрации 0. В обоих случаях удельная электропроводность слишком мала.
Рис.1-2.Зависимость эквивалентной
электропроводности от концентрации
Наибольшая электропроводность наблюдается в тех случаях, когда произведение C наибольшее (уравнение 1.2) [1].
Эквивалентная электропроводность
Она численно равна удельной электропроводности раствора, заключенного между двумя параллельными электродами, отстоящими друг от друга на расстоянии 1 см и имеющими такую площадь, что объем содержит 1 г-экв. растворенного вещества.
m = 1000/С, m =z, [m]=Cm*м2/моль [1.3]
С уменьшением концентрации эквивалентная электропроводность увеличивается и достигает предельного значения. Предельное значение эквивалентной электропроводности называется электропроводностью при бесконечном разбавлении и обозначается λ∞Факторы, влияющие на эквивалентную электропроводность, оказывают такое же влияние, как и на удельную электропроводность (рис.1-2).
λ ∞= λ∞К+ λ∞А, [1.4]
где λ∞К и λ∞А- предельные подвижности ионов, т.е. количество электричества, переносимое одной молекулярной массой эквивалента иона в одну секунду. Это соотношение называется законом независимого движения ионов: в бесконечно разбавленном растворе ионы движутся независимо друг от друга.[2].
Отношение эквивалентной электропроводности к предельной выражается уравнением:
λ / λ∞=α*( λК+λА)/( λК ∞+ λ А∞)= α fλ [1.5]
где fλ-коэффициент электрической проводимости.
Коэффициент электрической проводимости характеризует силы межионного взаимодействия, которые растут вместе с концентрацией ионов в растворе. Поэтому для слабых электролитов λ / λ∞= α, так как fλ =1(концентрация ионов не велика, следовательно, силы взаимодействия пренебрежимо малы), а для сильных электролитов λ / λ∞= fλ, так как α=1 (сильные электролиты практически полностью диссоциированы).
1.2.Теория Дебая-Онзагера
Эквивалентная электропроводность растворов сильных электролитов не должна зависеть от концентрации, так как концентрация не входит в уравнение 1.5, а =1. Однако это не подтверждается на практике. Зависимость эквивалентной электропроводности от концентрации может быть объяснена только с позиции теории Дебая-Онзагера. Согласно этой теории уменьшение эквивалентной электропроводности с увеличением концентрации растворов сильных электролитов связано с появлением двух эффектов: релаксационного и электрофоретического. Согласно теории в растворах сформировывается ионная атмосфера: каждый ион окружает себя ионами противоположного знака за счет сил притяжения разноимённых и отталкивания одноимённых зарядов. Формированию ионной атмосферы мешает броуновское (тепловое) движение. Поэтому возникающие в растворе тормозящие эффекты, которые связаны с формированием ионной атмосферы, находятся в зависимости от соотношения двух энергий: энергии броуновского движения частиц и потенциальной энергии притяжения.
Электрофоретическим эффектом называется торможение движения ионов при наложении электрического поля, вызванное перемещением иона в сторону, противоположную перемещению его ионной атмосферы. Окружающая ион атмосфера должна распадаться позади и образовываться впереди. Однако, она не успевает образовываться впереди и распадаться позади, поэтому количество зарядов противоположного знака позади иона больше чем спереди. Это – так называемое релаксационное торможение.
Согласно теории:
λ= λ∞-А.
 [1.6]
[1.6]где С-концентрация электролита, причём уравнение справедливо для разбавленных растворов (выше 0,002 н), А – константа, зависящая от природы растворителя т валентного типа электролита.
С повышением температуры электропроводность возрастает вследствие увеличения скорости движения ионов (уменьшается вязкость среды), следуя линейной зависимости:
λt=λ18(1+γ*(t-18)), [1.7]
где λt и λ18 эквивалентные электропроводности при температуре t и 180С.[3]
1.3. Аномальная подвижность ионов гидроксония и гидроксила
Ионы гидроксония и гидроксила обладают наиболее высокой подвижностью, так как они перемещаются двумя путями:
- За счет миграции, то есть движения ионов в направлении электрического поля.
- Эстафетный способ передачи ионов (Рис. 1-3.).
Подвижность иона гидроксония больше чем иона гидроксила, так как отрыв иона водорода от гидроксония требует меньшей энергии, чем от воды.

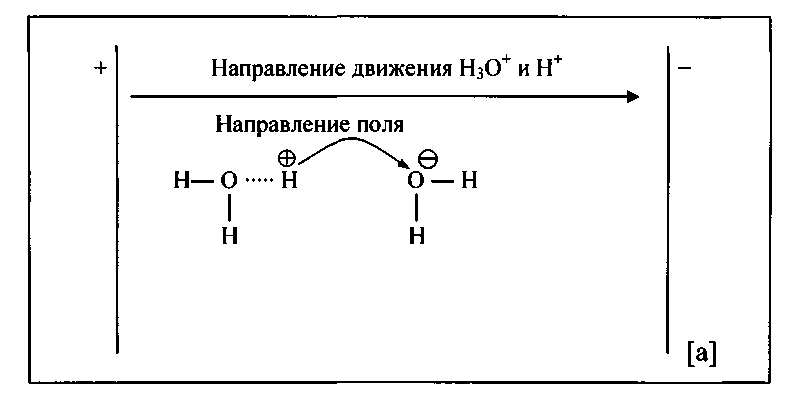
Рис. 1-3. Схема переноса катиона водорода (а) и гидроксид-иона (б)
1.4. Определение степени и константы диссоциации слабого электролита
по электрической проводимости раствора
Константу можно найти по закону Оствальда: Кд=Сα2/(1-α) [1.8]
а α можно найти по формуле:
α= λ / λ∞, а λ∞= λ∞К+ λ∞А.
П
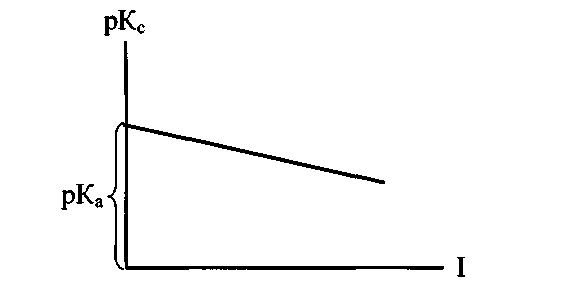 редельные подвижности λ∞К и λ∞А находят по справочникам.
редельные подвижности λ∞К и λ∞А находят по справочникам.Для определения термодинамической константы готовят растворы в широком интервале концентрации (от 0,001 до 0,05 моль/литр). Затем строят график pK=f (I).
Рис. 1-4. Зависимость константы диссоциации
от ионной силы раствора
1.5. Определение эквивалентной электропроводности слабого электролита при бесконечном разведении. Расчет константы диссоциации по методу Фуосса и Брея
Согласно этому методу уравнение Кд=сλ2/(λ∞( λ∞- λ)) преобразуется в уравнение: χ*10-3=Кс(λ∞)2*(1/ λ)-Ксλ∞ [1.9]
Температура при опыте поддерживается постоянная, поэтому Кс и λ∞.Затем составляется график χ=f(1/ λ).
Если Ксλ∞=α, то Кс(λ∞)2=tgα:
χ*10-3 = tgα*(1/ λ)- α. [2] [1.10]
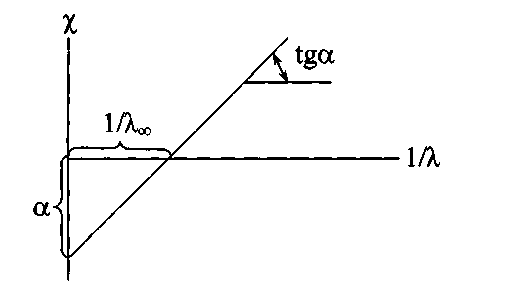
Рис. 1 -5. Определение константы диссоциации и предельной эквивалентной электропроводности слабого электролита по Фуоссу и Брэю
Кс= tgα/(λ∞)2.1.11]
2.Экспериментальная часть
2.1.Методика измерения электропроводности
Измерение электропроводности растворов электролитов практически сводится к измерению их сопротивления. Измерение сопротивления растворов может быть проведено как с помощью постоянного, так и с помощью переменного тока. На практике преимущественное распространение получил метод, основанный на применении переменного тока, так как это позволяет устранить поляризационное сопротивление.
Важное значение принадлежит устройству сосудов для измерения электропроводности.
Сопротивление раствора зависит не только от концентрации электролита, площади электродов и расстояния между ними, но и от их взаимного расположения и объёма раствора, так как в переносе электричества участвует значительно больший объем, чем тот, который находится между электродами. Электроды должны быть жёстко закреплены, поэтому их впаивают в стеклянные сосуды. Объём раствора, который заливается в сосуд, тоже должен быть постоянным. Если расстояние между электродами равно l, S-площадь их поверхности и f-фактор, зависящий от геометрических особенностей сосуда, то удельная электропроводность может быть рассчитана по формуле:
R= (1/χ)*f*(l/S),
где R измеренная величина сопротивления. Так как f, l, S-постоянны, то можно положить:
χ=K/R; K=f*l/S. K называют константой сосуда. [3]
Определение константы диссоциации слабого электролита
Приборы и материалы:
1.Щавелевая, янтарная и яблочная кислоты.
2.Установка для измерения электропроводности.
3.Темостат.
4.Сосуд для измерения электропроводности.
5.Набор мерной посуды.
В мерной колбе на 250мл готовят 0,01М раствор дикарбоновой кислоты (навеска берётся на аналитических весах). Сосуд для измерения электропроводности промывают дистиллированной водой, затем заполняют так, чтобы электроды полностью были погружены в раствор (в нашем случае 40мл).Проводят измерение удельной электропроводности. Затем разбавляют раствор вдвое. Снова определяют удельную электропроводность. Разбавление растворов повторяли 6-8 раз, затем рассчитывают удельную электропроводность и степень диссоциации для всей серии растворов.
Электропроводность измеряли при 180С. Константу ячейки измеряли по электропроводности стандартного раствора KCl, величина её в нашем случае составила 3,625*10-2 [5].
Определение λ∞и коэффициента электрической проводимости
раствора сильного электролита
fλ находят из отношения λ/λ∞ .Значение λ∞определяют экстраполяцией линейного участка зависимости λ=f(C0.5) C0.5=0. Находят λ∞как отрезок на оси ординат. Затем для каждого раствора рассчитывают fλ, строят зависимость fλ=f(C) [2].
2.2.Кондуктометрическое титрование
Кондуктометрическим титрованием называют метод определения концентрации или содержания вещества по кондуктометрическим кривым титрования, которые получают многократным измерением электрической проводимости после каждого прибавления небольшой порции титранта (0,1-0,2 мл) к титруемому раствору, находящемуся в кондуктометрической ячейке. Построив график в координатах 1/R объем титранта (VТ), называемый кондуктограммой, находят на нем точки пересечения отрезков прямых линий, соответствующих конечным точкам титрования (к.т.т.). По объему титранта (Vк.т.т.) пошедшего на титрование к.т.т., рассчитывают количество вещества в анализируемом растворе.
Кондуктометрическое определение количества вещества возможно потому, что при взаимодействии титруемого вещества с титрующим раствором изменяются ионная концентрация раствора и ионный состав. Ионы титруемого вещества заменяются эквивалентным количеством ионов титранта, имеющих иную подвижность. Отрезки на кондуктограмме получаются прямолинейными, если реакция между определяемым веществом и титрантом необратима и проходит стехиометрично. Обратимость реакции из-за гидролиза образующейся соли, растворения осадка или диссоциации комплексов приводит к появлению округлений вблизи к.т.т. и уменьшению прямолинейных отрезков кондуктограммы. Вследствие этого повышаются ошибки титрования. Вследствие увеличения концентрации титранта в 25 -50 раз по сравнению с концентрацией определяемого раствора уменьшается разбавление, а в связи с этим уменьшается искажение кондуктограммы. Кондуктометрическое титрование требует больше времени, чем титрование с цветными индикаторами, поэтому применяют его тогда, когда индикаторное титрование невозможно:
1) при определении концентрации очень слабых кислот и оснований;
2) при анализе окрашенных растворов или растворов с осадком;
3) при анализе многокомпонентных систем.
Кондуктометрическое титрование расширяет возможности титрометрического метода анализа и создает условия, при которых нельзя перетитровать раствор, как при индикаторном титровании.
Титрование слабой кислоты сильным основанием
Согласно уравнению:
χ=С*α(λК+λА)*103
и реакции
HA+NaOH=H2O+NaA
удельная электропроводность исходного раствора(HA), раствора в к.т.т.(NaA) и перетитрованного раствора (NaA,NaOH)выражаются уравнениями:
χИ*10-3=СHA*α(λH+λA);
χк.т.т.*10-3= СHA(λH++λA-);
χп*10-3= СNaA(λH++λA-)+ СNaOH(λNa++λA-);
При титровании до к.т.т. χ изменяется за счёт изменения состава ионов(ионы водорода заменяются на Na+ ) и изменения концентрации ионов(вместо мало диссоциированной кислоты образуется полностью диссоциирующая соль).За счёт первого фактора χ уменьшается, за счёт второго χ растёт [5].
3.Обсуждение результатов
3.1.Титрование щавелевой кислоты
Среди незамещённых дикарбоновых кислот щавелевая кислота является наиболее сильной (табл. 3-1).
Таблица 3-1. Константы диссоциации дикарбоновых органических кислот.
| Кислота | К1 | К2 |
| Щавелевая | 5,4.10-2 | 5,4.10-5 |
| Яблочная | 3,5.10-4 | 8,9.10-6 |
| Янтарная | 6,2.10-5 | 2,3.10-6 |
Значит, она будет диссоциирована по первой стадии в значительной степени в разбавленных растворах (0,01М):
H2C2O4=H++HC2O4-
В тоже время не значительно по второй:
HC2O4-=H++C2O42-
Это связано с тем, что pK2 относительно мало, и с тем, что диссоциация подавляется относительно высокой концентрацией ионов водорода, созданной за счёт первой стадии.
Титрование гидроксидом калия
Кривая титрования представлена на рис. 1-1. Из графика видно, что электропроводность вначале уменьшается (до первой точки эквивалентности). Это объясняется заменой ионов водорода на менее подвижные ионы калия, несмотря на образование, полностью диссоциирующей соли. Однако вклад последнего фактора в общий ход кривой не значителен. Его влияние проявляется близ первой точки эквивалентности, что и объясняет появление округлений. В процессе титрования до первой точки эквивалентности происходила нейтрализация щавелевой кислоты до гидроаксалат-иона. Затем происходит рост удельной электропроводности и нейтрализация гидроаксалат-иона до второй точки эквивалентности. Это объясняется увеличением количества оксалат-анионов и катионов титранта. После второй точки эквивалентности происходит резкое увеличение электропроводности. Это объясняется возрастанием концентрации гидроксид-ионов, обладающих аномально высокой подвижностью.
Вторая точка эквивалентности может быть ярко выражена только в том случае, если подвижности аниона кислоты и гидроксила будут резко отличаться. В противном случае изгиб будет незаметен, и трудно обнаружить вторую точку эквивалентности по графику. В таком случае найти вторую точку эквивалентности можно, зная, что количество щёлочи, затраченное на первую точку эквивалентности равно количеству щёлочи, затраченной на вторую точку эквивалентности. Гипотетическую первую точку эквивалентности, не имеющую аналитического значения, можно найти экстраполяцией линейных участков кривых.
Таблица 3-2. Предельная эквивалентная электропроводность (подвижность) ионов в водных растворах при 25 °С
| Ион | Подвижность |
| H+ | 349,8 |
| NH4+ | 73,6 |
| |K+ | 73,5 |
| Na+ | 50,1 |
| CH3NH3+ | 58,7 |
| ОН– | 198,3 |
| 1/2С2О42 – | 74,0 |
| 1/2СО32 – | 69,3 |
| НСО3– | 54,6 |
| СНзСОО– | 35,8 |
Титрование гидроксидом аммония
Титрование гидроксидом аммония является более точным, чем титрование гидроксидом калия. Ход кривой практически совпадает с ходом кривой, полученной при титровании гидроксидом калия, до второй точки эквивалентности. Несмотря на то, что гидроксид аммония – слабое основание. Но ведь продукты аналогичны продуктам, образующимся при титровании гидроксидом калия:
HA-+H++NH4OH=NH4++HA-+H2O
HA-+ NH4OH =A2-+H2O+ NH4+
График титрования гидроксидом аммония показан на рисунке 1.1.Сравнив два графика, показанные в одной системе координат, видно, что они расходятся только после второй точки эквивалентности (кривая титрования гидроксидом аммония далее идёт горизонтально). Это объясняется очень близкими подвижностями иона аммония и калия. Расхождение графиков объясняется тем, что гидроксид аммония слабый электролит и не влияет на электропроводность раствора после полной нейтрализации кислоты. Тем более диссоциация электролита подавляется высокой концентрацией ионов аммония, образовавшихся при титровании кислоты.
3.2. Титрование янтарной кислоты
Кривые титрования янтарной кислоты сильными основаниями представлены на рисунке 1-2. Они имеют существенное отличие от зависимостей, полученных для щавелевой кислоты. Для относительно разбавленных растворов янтарной кислоты наблюдается первоначальное незначительное понижение электропроводности, и на кривой появляется минимум, не имеющий аналитического значения. Он не соответствует первой точке эквивалентности, которая расположена на восходящем участке кривой. Для более концентрированных растворов понижение электропроводности может отсутствовать вовсе, а кривая состоит из двух участков, на которых происходит увеличение электропроводности раствора. Особенности данных кривых титрования можно объяснить более низкими значениями, как первой, так и второй констант кислотности янтарной кислоты по сравнению со щавелевой кислотой (табл. 3-1).
Более низкое значение констант кислотности янтарной кислоты по сравнению со щавелевой объясняется теорией электронных смещений, достаточно подробно изложенной в целом ряде книг [9-13]. Следует заметить, что только в щавелевой кислоте наблюдается образование единой сопряженной π-системы между двумя карбоксильными группами, в остальных дикарбоновых кислотах возможно проявление лишь индукционного эффекта, передающегося через систему σ-связей. Влияние индукционного фактора проявляется значительно слабее, при этом происходит затухание индукционного эффекта по мере удаления двух карбоксильных групп друг относительно друга, что и приводит к низкому значению первой константы кислотности для янтарной кислоты. Следует также учесть маленькую разность рК2— рК1, которая в случае янтарной кислоты составляет 1,43, то есть меньше 2. Дифференцированное титрование в таком случае было бы возможным, если бы подвижности ионов НА - и А2- сильно отличались между собой. В справочных данных по подвижностям гидросукцинат-аниона и сукцинат-аниона мы не нашли, однако для угольной кислоты подвижности карбонат-иона и гидрокарбонат-иона различаются незначительно (табл. 3-2).
Кроме этого следует учесть и тот факт, что впервой точке эквивалентности доля гидросукцинат-иона едва достигает 70% (рис. 2-2), в то время, как для щавелевой кислоты доля гидроаксалат-иона в аналогичной ситуации достигает 95%(рис.2-1). В действительности мы можем наблюдать излом только во второй точке эквивалентности (рис 1-3).
Титрование слабым основанием представлено на рисунке 1-3. И в этом случае вид кривой свидетельствует том, что определение первой точки эквивалентности невозможно, однако излом в конечной точке титрования может быть определён с высокой степенью надёжности.
3.2.Титрование яблочной кислоты
Яблочная кислота является более сильной, чем янтарная, но более слабой, чем щавелевая кислота (табл. 1-1). Это объясняется тем, что отсутствует сопряжение между двумя карбоксильными группами. В тоже время она сильней, чем янтарная, так как присутствует отрицательный индуктивный эффект за счёт гидроксильной группы. Кривые титрования кислоты сильным и слабым основанием представлены на рисунке 1-3. Они имеют сглаженную нелинейную форму, что практически не позволяет установить первую точку эквивалентности – на кривой титрования наблюдается минимум, не имеющий аналитического значения. При титровании сильным основанием затруднительно определение и второй точки эквивалентности.
Вначале наблюдается небольшой изгиб. Но этот первый изгиб не соответствует первой точке эквивалентности. Уменьшение электропроводности происходит благодаря тому, что ионы водорода заменяются на менее подвижные катионы калия или аммония, несмотря на то, что образуется полностью диссоциирущая соль. Влияние этого фактора начинает сказываться только близ первого изгиба, поэтому электропроводность начинает “рано” повышаться. Вторая точка эквивалентности практически не видна (видимо, подвижности гидроксид-иона и гидромалат-иона близки). После изгиба кривая идёт вверх (удельная электропроводность повышается). Это объясняется тем, что ионы яблочной кислоты заменяются на аномально подвижные ионы гидроксила.
При титровании гидроксидом аммония наблюдается аналогичный ход кривой, но в отличие от кривой титрования гидроксидом калия, после второй точки эквивалентности удельная электропроводность не изменяется.
Замена сильных оснований слабыми наглядно демонстрирует преимущества в определении второй точки эквивалентности, излом в которой достаточно определён.
Таким образом, можно заключить, что в случае относительно сильных кислот с pK2-pK 1≥3 можно осуществить дифференцированное титрование с независимым определением двух точек эквивалентности с использованием в качестве титранта как сильных, так и слабых оснований. Для очень слабых кислот с pK2-pK 1<5 определение первой точки эквивалентности невозможно, однако вторая точка эквивалентности определяется с достаточной степенью надёжности при титровании как сильными, так и слабыми основаниями. Для кислот с промежуточным значением константы кислотности использование сильных оснований не даёт возможности надёжно определить ни одну из точек эквивалентности. И в этом случае применение слабого основания оказывается более эффективным с аналитической точки зрения.
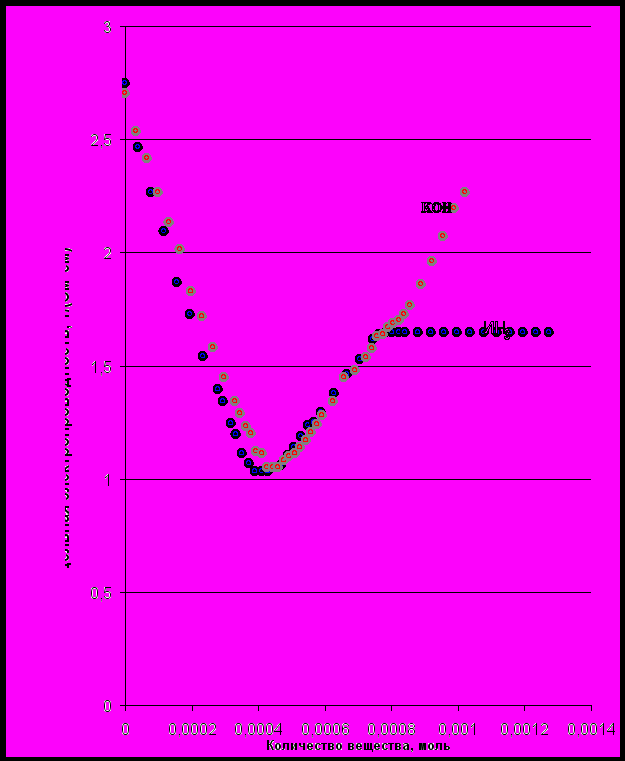
Рис. 1-1. Кривые титрования щавелевой кислоты сильным и слабым основанием
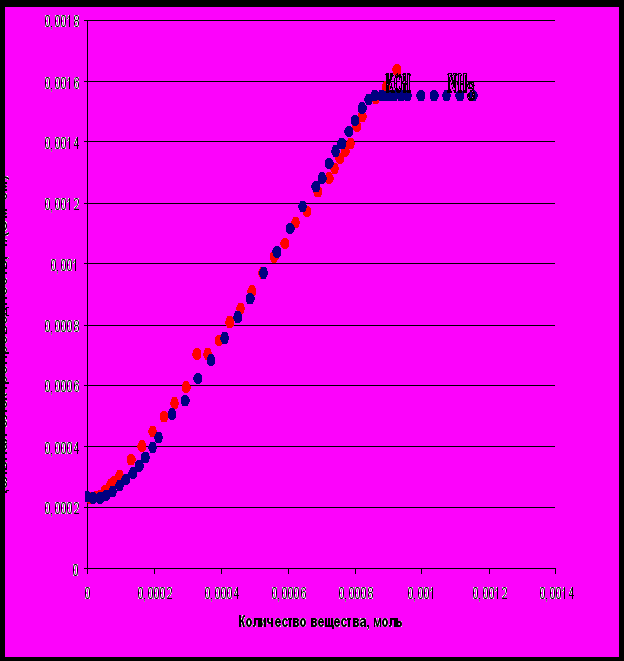
Рис. 1-2. Кривые титрования янтарной кислоты сильным и слабым основанием

Рис. 1-3. Кривые титрования яблочной кислоты сильным и слабым основанием
Рис. 2-1. Доля частиц в зависимости от pН раствора щавелевой кислоты
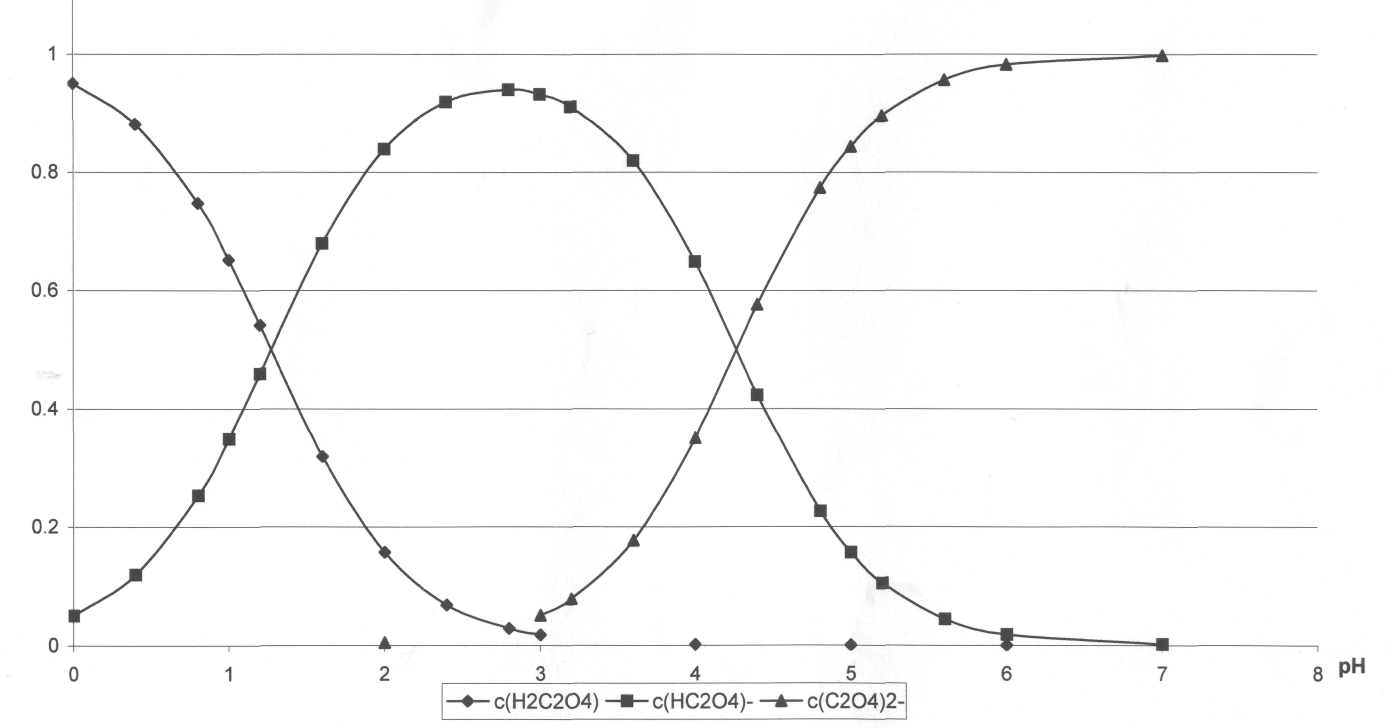
Рис. 2-2. Доля частиц в зависимости от pH раствора щавелевой кислоты
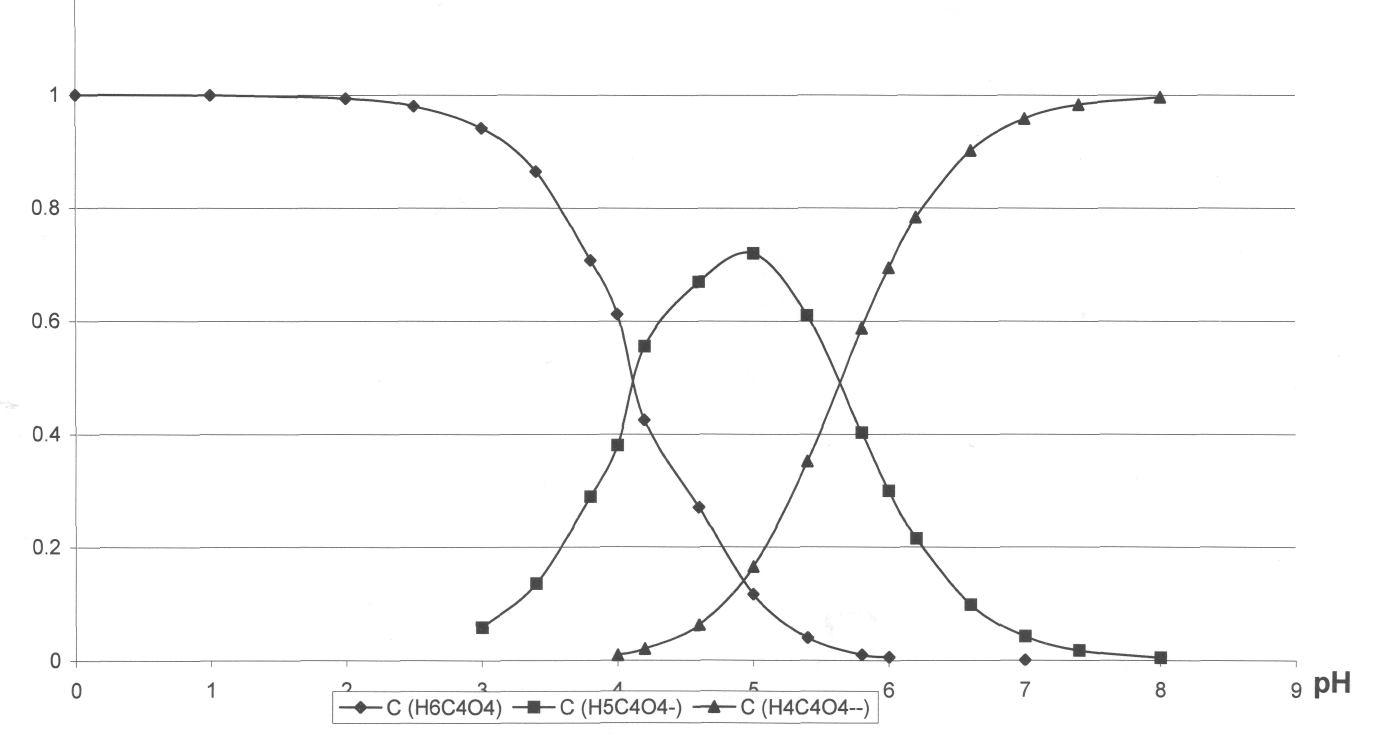
Выводы
1. Показана возможность дифференцированного титрования щавелевой кислоты как слабыми, так и сильными основаниями.
2. Для кислот, у которых pK>5, показано, что дифференцированное титрование провести не удаётся, но можно определить вторую точку эквивалентности как при титровании сильными, так и слабыми основаниями.
3. Кислоты, имеющие промежуточное значение 2
Литература
1.Шаталов А.Я., Маршаков И.К. Практикум по физической химии. М.:Высшая школа, 1968.
2. Ермаков А.И., Арасимович В.В., Смирнова-Иконникова М.И., Мури И.К. Методы биохимического исследования растений. М-Л.: СЕЛЬХОЗГИЗ, 1952.
3.Практикум по электрохимии. Под ред. Б.Б. Дамаскина. М.: Высшая школа,1991.
4.Евстратова А.И., Купина Н.А., Малахова Е.Е. Физическая и коллоидная химия. М.: Высшая школа, 1990.
5.Левин А.И., Помосов А.В. Лабораторный практикум по теоретической электрохимии. М.: Металлургия, 1996.
6.Астафуров В.И. Основы химического анализа. М.: Просвещение, 1982.
7.Воскресенский П.И., Неймарк А.М. Основы химического анализа. М.:Просвещение, 1971.
8.Чудякова Т.А., Крешков А.П.Кондукеометрический метод анализа. М.: Высшая школа, 1975.
9.Марч Дж. Органическая химия. М.: Мир, 1987. – Т.1-4.
10.Моррисон Р., Бойд Р. Органическая химия. М.: Мир, 1974.
11.Несмеянов А.Н., Несмеянов Н.А.Начала органической химии. М.: Химия, 1974. – Т 2.
12.Общая органическая химия/ Под редакцией Д.Бартона и В.Д.Оллиса. М.: Химия, 1982. – Т 2.
13.Пальм В.А. Введение в теоретическую органическую химию. М.: Высшая школа, 1974.