
Научные основы прогноза токсичности и опасности химических веществ с учетом механизма токсического действия. 14. 02. 01 Гигиена
| Вид материала | Автореферат |
- Отравление аварийными химически опасными веществами, 33.95kb.
- Токсикомания шаг в сторону наркомании, 29.31kb.
- Показатели безопасности изделий регламентируются с учетом функционального назначения,, 184.97kb.
- Вопрос Параметры звука, 183.33kb.
- Волощенко О. И., Медяник, 2522.04kb.
- Программа спецкурса «методы расчета физико-химических свойств веществ» для студентов, 40.47kb.
- Т е м ылекций по гигиене для студентов IV курса медицинского факультета по специальностям, 21.78kb.
- Технология переработки отходов производств, использующих высокочистый кремний 05. 17., 277.13kb.
- О государственной регистрации потенциально опасных химических и биологических веществ, 360.85kb.
- «О пользе молока», 74.05kb.
Ключевая реакция биотрансформации определялась по результатам расчета наиболее стабильного радикала, образующегося при отрыве атома водорода от углерода. В процессе дальнейшей биотрансформации происходит присоединение гидроксильного радикала и образование карбонильных метаболитов с отрывом НCl или HBr. Структура конечного метаболита однозначно определяется структурой радикала, образующегося на первой стадии. Для обучающей выборки, включающей 19 веществ и тестовой выборки, включающей 8 веществ, проведен прогноз ключевой реакции биотрансформации, т.е. методом АМ1 рассчитаны минимальные значения энтальпии активации модельной реакции образования радикалов (На#) (Yin H. et al., 1995). По результатам расчетов установлено граничное значение этого параметра - 26 ккал/моль. Если энтальпия активации превышает эту величину, вещества не подвергаются биотрансформации по пути окислительного гидроксилирования и не обладают канцерогенной активностью.
Для метаболитов, соответствующих ключевой реакции биотрансформации рассчитан параметр, характеризующий реакционную способность в реакции с ДНК – энергия нижней свободной молекулярной орбитали, разрыхляющей по карбонильной связи С=О (ЕМО). Для характеристики выраженности канцерогенного эффекта для соединений обучающей выборки с использованием информации из базы данных CPDB были рассчитаны значения канцерогенного потенциала lg(M/TD50). Соединения обучающей выборки было отнесены к определенному классу по канцерогенности в соответствии с предложенной классификацией.
В результате компьютерного эксперимента был осуществлен правильный прогноз канцерогенной активности известных канцерогенов трихлорметана и 1,2,3-трихлорпропана, сделан прогноз класса опасности 8 неизученных соединений. Результаты (Таблица 11) не противоречат оценке, данной экспертами (Woo Y.-T. et al., 2002).
По результатам прогноза 1,2-дибром-1,1,5-трихлорпентан может обладать канцерогенной активностью, в настоящее время ПДК установлена по органолептическому признаку вредности (запах).
Таблица 11. Прогноз класса опасности галогенированных алканов по канцерогенному эффекту
Название | Метаболит | Н# min (ккал/ моль) | ЕНСМОметабо-лита (эВ) | Прогноз класса опасности |
| 1,2,3-трихлорпропан | 2-хлорпропеналь | 23,31 | -0,469 | 1 |
| 1,1,1,2-тетрабром-2-хлорэтан | 1,1,1-бромацетилхлорид | 23,36 | -0,548 | 2 |
| 2,3-дихлорбутан | 3-хлорбутан-2-он | 22,17 | 0,204 | 3 |
| 1,1,5,5-тетрахлорпентан | 1,1-дихлорпентаноил хлорид | 24,94 | -0,301 | 3 |
1-хлороктан | Октаналь | 22,86 | 0,888 | 4 |
2-хлордодекан | додека-2-он | 22,53 | 0,779 | 4 |
| 1,2-дихлор-2-метилбутан | 2-хлор-2-метилбутаналь | 23,15 | 0,235 | 3 |
1,2-дибром-1,1,5-трихлорпентан | 1,2-дибром-1,1-дихлорпента-наль | 23,54 | -0,376 | 2/3 |
1,2-дибромпропан | Бромацетон | 22,34 | -0,115 | 2 |
Обоснована необходимость пересмотра норматива в воде для 1,2,3-трихлорпропана – вероятного канцерогена для человека по классификации МАИР (группа 2А). В настоящее время 1,2,3-трихлорпропан нормирован по органолептическому признаку вредности.
При изучении канцерогенной активности ароматических аминов исходили из гипотезы, что ключевой реакцией биотрансформации является перенос электрона с субстрата на фермент с образованием катион-радикала (Franke R. et al., 2001) R. В обучающую выборку из 82 соединений включены аминопроизводные бензола, флуорена, нафталина, бифенила. В результате ДСМ-эксперимента на этом массиве были порождены гипотезы, включающие как структурную часть, описанную с помощью графов, так и интервалы значений числовых параметров – энергии высшей заполненной молекулярной орбитали и логарифма коэффициента распределения октанол/вода. С использованием порожденных гипотез все соединения обучающей выборки были правильно отнесены к канцерогенным и не канцерогенным веществам (выполнен критерий достаточного основания). Был сделан прогноз для трех неизученных ариламинов. Согласно прогнозу, 3,5-диметиланилин, 3,5-диметоксианилин и 5-хлор-орто-анизол не должны обладать канцерогенной активностью. В тестовую выборку были включены о-метиланилин, п-метиланилин и м-метиланилин. По результатам прогноза о-метиланилин и п-метиланилин должны обладать канцерогенной активностью, а м-метиланилин не активен, что согласуется с экспериментальными данными.
В целом результаты прогноза канцерогенной активности свидетельствуют, что использование числовых параметров позволяет существенно повысить как полноту (количество тестовых соединений, для которых с помощью сгенерированных гипотез удается прогнозировать класс по канцерогенности), так и точность ДСМ-прогноза.
Прогноз порядка величины безопасных уровней канцерогенных веществ в воде
Использованное при прогнозе показателей токсикометрии разделение на классы не дает возможности прогнозировать безопасные уровни. Поэтому была поставлена задача разработки классификации для их прогноза. Проанализированы величины безопасных уровней веществ, для которых ПДК в воде установлены с учетом канцерогенного эффекта. Величины ПДК в основном различаются на порядки. Эта же закономерность – различие на порядки характерна и для критериев, используемых для оценки риска канцерогенных веществ. В связи с этим, при разработке классификации опасности канцерогенов для цели прогноза их безопасных уровней в воде можно принять, что необходимым и достаточным является прогноз порядка величины. Такая классификация должна быть основана на величинах наименьших доз канцерогенных веществ, определенных в эксперименте. Для нескольких сот веществ имеется информация о величинах LTD10 – нижней доверительной границы дозы, соответствующей возникновению рака у 10% животных. Именно эти величины были использованы для классификации. Для большинства веществ наблюдается закономерность: чем ниже величина LTD10, тем ниже величина ПДК. Однако по зависимости величин ПДК от LTD10 интервалы значений ПДК, соответствующие высокой, умеренной и слабой канцерогенной активности определить не удается. Кроме того, классификация должна учитывать информацию о канцерогенной опасности не только для лабораторных животных, но и для человека.
Таблица 12. Алгоритм определения класса веществ по канцерогенной опасности в соответствии с величиной LTD10 c учетом классификации МАИР
| группа по классификации МАИР) | Значения LTD10 (мг/кг) | |||
| <0,01 | 0,01-1 | 1-10 | >10 | |
| Класс опасности | ||||
| 1 | 1 | 1 | 2 | |
| 2A | 2 | 2 | 3 | |
| 2B | 2 | 3 | 3 | 3 |
| 3 | 3 | 3 | 4 | 4 |
Высказано предположение, что объединение классификации по LTD10 и классификации канцерогенности для человека, принятой МАИР, позволит более правильно предсказывать безопасные уровни.
Такой подход аналогичен принятому ЕРА для оценки сравнительной опасности канцерогенов. Алгоритм классификации представлен в таблице 12.
Можно видеть (рис. 6), что различным классам в основном соответствуют различные интервалы значений ПДК. Для веществ 3 и 4 классов характерны значения ПДК выше 0,01 мг/л, для веществ 2-го класса – от 0,0001 до 0,01 мг/л, для веществ 1 класса – ниже 0,0001 мг/л.
Класс опасности по предлагаемой классификации для неизученных соединений может быть предсказан по результатам системы прогноза.
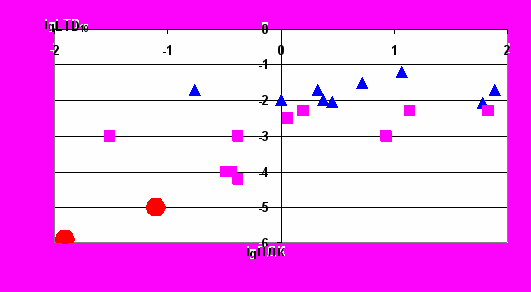
 - 1 класс опасности
- 1 класс опасности - 2 класс опасности
- 2 класс опасности - 3 класс опасности
- 3 класс опасности Рисунок 6. Зависимость ПДК веществ, нормированных в воде по канцерогенному эффекту, от LTD10 c указанием класса опасности
После установления класса опасности неизученного вещества порядок величины ПДК может быть предсказан в соответствии с таблицей 13.
Таблица 13. Предлагаемые интервалы значений безопасных уровней в соответствии с классами опасности по канцерогенному эффекту
| Класс канцерогенной опасности | Интервалы значений безопасных уровней (мг/л) |
| 1 класс – высоко опасные вещества | ПДК< 0,0001 |
| 2 класс – опасные вещества | 0,0001 ПДК < 0,01 |
| 3 класс –умеренно опасные вещества | 0,01 ПДК<0,1 |
| 4 класс – мало опасные вещества | Канцерогенный эффект не прогнозируется |
В таблице 14 приведены вещества, нормативы которых в воде предлагается скорректировать или вновь принять по результатам системного прогноза и величины действующих нормативов.
Выделены вещества, для которых установлены или рекомендованы нормативы в воде за рубежом. Порядки величин, определенные по предлагаемой методике, совпали с порядками этих безопасных уровней.
Разработанные методы прогноза позволили определить классы опасности по канцерогенному эффекту 35 веществ, дать рекомендации по корректировке ПДК 11 веществ. Рекомендованы порядки безопасных уровней 4-х ранее не изученных не нормированных веществ без проведения токсикологических экспериментов на животных.
Таким образом, предлагаемая методика прогноза порядка величины безопасных уровней веществ позволяет скорректировать нормативы веществ, которые были установлены без учета канцерогенного эффекта, а также предсказать порядок величины норматива неизученных веществ, которые по результатам прогноза канцерогенности могут обладать этим видом эффекта.
Таблица 14. Порядки ПДК канцерогенных веществ в воде, предлагаемые на
основании прогноза
| Вещество | Порядки ПДК | ПДК, мг/л | Лимитиру-ющий показатель вредности | Класс опасности |
| Акрилонитрил | 10 -3 | 2,0 | с.-т. | 2 |
| Бензил хлористый | 10 -3 | 0,001 | с.-т. | 2 |
| Бензотрихлорид | 10 -3 | - | - | - |
| Пропиленоксид | 10 -2 | 0,01 | с.-т. | 2 |
| 1,2,3-Трихлорпропан | 10 -4 | 0,07 | орг. зап. | 3 |
| Гексахлорциклогексан | 10 -4 | 0,02 | орг. зап. | 4 |
| Гептахлор | 10 -4 | 0,05 | с.-т. | 2 |
| Гидразин* | 10 -2 | 0,01 | с.-т. | 2 |
| Катехол | 10 -2 | 0,1 | орг. окр. | 4 |
| o-Нитроанизол | 10 -2 | 0,3 | орг.привк. | 3 |
| Бензидин** | <10 -4 | - | - | - |
| 4-Аминобифенил** | <10 -4 | - | - | - |
| 2-метиланилин гидрохлорид** | 10-3 | - | - | - |
| 4-метиланилин | 10 -2 | 0,6 | орг. окр. | 3 |
* - ОДУ
** - вещество ранее в воде нормировано не было
Комплексная система, основанная на совмещении логико-комбинаторного ДСМ-метода и анализа числовых параметров соотношений структура-биотрансформация-токсичность
На основании проведенных исследований была разработана комплексная версия интеллектуальной ДСМ системы, в которой стандартная система дополнена квантово-химическим модулем генерации метаболитов и расчета их электронных параметров, а в ДСМ-рассуждения включен анализ числовых параметров. Таким образом, комплексная система основана на совмещении логико-комбинаторных методов и методов квантовой химии, используемых для моделирования процессов взаимодействия веществ с организмом, и в частности процессов биотрансформации под действием ферментных систем.
Разработано также сопряжение ДСМ-системы с созданной нами базой данных по токсичности и опасности веществ в воде WATERTOX и базой данных по канцерогенности CPDB, что дает возможность формировать обучающую выборку для прогноза класса опасности по острой и хронической токсичности, а также класса опасности по канцерогенному эффекту. В основу структуры базы данных WATERTOX легли показатели разработанной ранее (Красовский Г.Н., Жолдакова З.И. и соавт., 1992) формы аннотационных карт опасности химических веществ. Информацию по веществу можно разделить на следующие блоки:
- блок идентификационной информации, содержащий название вещества, синонимы и товарные наименования, регистрационные номера, в частности номер по CAS;
- блок информации о физико-химических свойствах вещества; блок информации, содержащий данные об органолептических свойствах и данные по влиянию на процессы самоочищения водоемов, блок информации по токсичности для гидробионтов и млекопитающих в острых, подострых и хронических опытах (смертельные, пороговые и безвредные дозы);
- блок информации по специфическим и отдаленным эффектам вещества (пороговые и максимальные недействующие дозы вещества по данному виду эффекта, класс по МАИР; информация по нормативам в объектах окружающей среды.
Для сопряжения базы данных WATERTOX с ДСМ системой был реализован ее перевод из DBF-формата (кодировка DOS) в MDB-формат (кодировка Windows).
Доступ к базе данных осуществляется через технологию Microsoft ADO, что позволяет реализовать ее использование в сети.
Создана программа для формирования файла свойств при создании выборки из базы данных WATERTOX. Набор свойств, в простейшем случае «+» и «-», характеризует отнесение соединения к определенному классу опасности. Предусмотрен прогноз класса опасности по острой токсичности (по величине ЛД50 для крыс и мышей) и хронической токсичности (по величине ПКхр и МНКхр.). Алгоритм системы приведен на рисунке 7.
Если прогноз проводился с использованием числовых параметров, проводится расчет этих параметров в соответствии с используемой моделью по программам, интегрированным в систему, и значение параметра добавляется к файлу параметров, а файл описания структуры – в директорию эксперимента. В случае, если соединение относится к неизученному ряду, подбирается обучающая выборка из баз данных по токсичности и опасности WATERTOX или канцерогенности (CPDBase) и осуществляется стандартный компьютерный ДСМ эксперимент с анализом сходства структур соединений. Развитие системы заключается в расширении количества структурных рядов и видов эффектов, для которых в соответствии с определяющей стадией механизма токсического действия разработаны модели прогноза структура-биотрансформация-токсичность и определены числовые параметры.
Для расчета электронных параметров соотношений структура-биотрансформация-токсичность разработано сопряжение ДСМ-системы и баз данных с квантово-химическими программами расчета электронных параметров. Для каждого вида эффекта и каждого структурного ряда параметры специфичны и определяются гипотезой о ключевой реакции взаимодействия вещества с организмом. Возможность использования того или иного квантово-химического метода также определяется спецификой структурного ряда. Метод Хюккеля интергрирован в ветвь системы, предназначенную для прогноза наличия и степени выраженности канцерогенного эффекта. Для расчета параметров соединений, не имеющих системы сопряженных связей, и в том случае, когда возможность биоактивации зависит от конформации молекулы вещества или его метаболитов, применяют полуэмпирические квантово-химические методы.

Рисунок 7. Алгоритм системы прогноза токсичности и опасности химических веществ.
Сопряжение с программой, реализующей полуэмпирический метод, проиллюстрировано на примере модели для прогноза канцерогенности галогенированных алканов. Согласно гипотезе о ключевой реакции биотрансформации соединений этого ряда и ее связи с канцерогенностью, для тестового соединения рассчитывается разность теплот образования радикалов, получающихся при отрыве атома водорода от всех неэквивалентных по симметрии атомов углерода молекулы и исходного соединения. Эта разность характеризует энтальпию активации. Минимальное значение энтальпии определяет положение биотрансформации. Гипотеза состоит также в том, что если это минимальное для данной молекулы значение больше 27 ккал/моль, соединение не подвергается метаболизму и не будет канцерогеном. В противном случае строится структура метаболита и рассчитывается энергия нижней свободной молекулярной орбитали метаболита. Разность теплот образования радикала и исходного соединения и энергия нижней свободной молекулярной орбитали метаболита используются в качестве параметров при ДСМ-эксперименте по прогнозу канцерогенности.
Расчеты производятся с использованием построенного в химическом редакторе файла описания структуры, затем проводится минимизация геометрии методом молекулярной механики и полуэмпирическим методом АМ1. Результирующее значение теплоты образования исходной молекулы, рассчитанное этим методом, запоминается. Затем моделируется отрыв атомов водорода путем соответствующего изменения файла описания структуры. Теплота образования получившихся радикалов рассчитывается неограниченным методом Хартри-Фока. Положение отрыва атома водорода, соответствующее минимальному значению энтальпии активации, однозначно определяет структуру карбонильного метаболита. Нахождение минимального значения энтальпии активации, построение структуры метаболита, соответствующего минимальной энергии активации, расчет энергии нижней свободной молекулярной орбитали метаболита и добавление параметров во входной файл ДСМ-системы происходят автоматически.
Результатом является прогноз значения свойств (+/-), что означает прогноз отнесения соединения к определенному классу по канцерогенности.
Система содержит результаты ДСМ-экспериментов для определенных структурных рядов и различных токсических эффектов, т.е. гипотезы, сгенерированные на обучающих выборках из соединений этих структурных рядов. В настоящее время разработаны и внесены в систему модели для прогноза класса канцерогенности, мутагенности, острой и хронической токсичности, метгемоглобинобразования.
Обсуждение результатов исследований.
Правомерность принципов, лежащих в основе разработанной системы прогноза токсичности и опасности химических веществ подтверждается тем, что в последние годы подходы, связанные с учетом биотрансформации при прогнозе токсичности, стали активно развиваться за рубежом. В связи с этим выделилось новое направление исследований, сокращенно называемое ADME (absorption, distribution, metabolism, excrection), т.е. изучение и прогноз абсорбции, распределения в организме, биотрансформации (метаболизма) и выделения химических веществ. Определяющая роль этих стадий взаимодействия вещества с организмом в развитии токсического эффекта подчеркивалась еще в 1980-х годах в работах З.И. Жолдаковой в ходе разработки патогенетической модели интоксикации (Жолдакова З.И., 1987).
Развиваемый нами подход к учету биотрансформации при прогнозе токсичности и опасности основан на некоторой модели (гипотезе) о механизме биотрансформации и квантово-химических расчетах, и не требует наличия обширных баз данных по метаболизму, как в случае прогноза биотрансформации с обучением на положительных и отрицательных примерах.
Основное положение нашего подхода – необходимость понимания механизма токсического действия на молекулярном уровне для построения соотношений структура-токсичность, разделяется в настоящее время большинством исследователей. Основным итогом данной работы является вывод о безусловной важности учета механизма токсического действия и процессов биотрансформации для прогноза токсичности и опасности химических соединений, и адекватности применения методов квантовой химии и логико-комбинаторного анализа для целей прогноза.
Апробация предложенной системы прогноза показала, что система позволяет прогнозировать вид и механизм токсического эффекта, выбирать адекватные показатели токсического действия и дозы для проведения острых и хронических экспериментов по обоснованию ПДК веществ, сокращать объем исследований при обосновании временных нормативов веществ в окружающей среде.