
Синтез, химические превращения биологически активных функционализованных ( O, n )гетеро-1,3-диенов и их кольчатых аналогов 02. 00. 03 органическая химия
| Вид материала | Автореферат |
- 2,4-диарилбицикло 1]нон-2-ен-9-оны: синтез, строение и некоторые химические превращения, 300.55kb.
- Синтез и свойства биологически активных соединений, содержащих no-донорный фрагмент, 577.21kb.
- Синтез и химические свойства дикарбонильных соединений адамантанового ряда 02. 00., 667.04kb.
- Рабочая программа по дисциплине ен. Ф. 04 «Органическая химия», 422.49kb.
- Рабочая программа по дисциплине ен. Ф. 04 «Органическая химия», 320.1kb.
- Новые органические лиганды n 2 s 2 -типа и их комплексные соединения с ni(II), Co(II),, 232.86kb.
- Синтез и превращения новых гетарилкарбаматов с индольным, 1,3-бензотиазол-2(3 н )-оновым, 279.88kb.
- Образовательная программа 240100 Химическая технология и биотехнология Дисциплина Химия, 54.66kb.
- Синтез и модификация 3a,4,5,9b-тетрагидро-3 н -циклопента[ c ]хинолинов, 408.36kb.
- Рабочая программа дисциплины (модуля) «математический анализ», 424.74kb.
Основное содержание работы
1. Синтез и особенности строения поликарбонильных систем со сближенными 1,2- и 1,3-дикарбонильными звеньями на основе активированных акцепторами окса-1,3-диенов
Как широко известные 1,3-дикарбонильные системы (ДКС), так и некоторые производные ДКС с сочленённым 1,2-дикарбонильным звеном – ацилпировиноградные кислоты (АПК) и лактоны их γ-енольной формы – фуран-2,3-дионы достаточно хорошо изучены. Нами впервые исследованы разнообразные поликарбонильные системы со сближенными 1,2- и 1,3-дикарбонильными звеньями – производные моно- и полиокса-1,3-диенов. Наиболее интересными как в синтетическом, так и структурном плане являются поликетиды на основе β-трикетонов с различными карбонильными акцепторами.
Известно, что поликетиды, имеющие β-трикетонное звено, успешно используются в органическом синтезе, в том числе в реакциях гетероциклизации. Доступными соединениями этого класса являются производные хелидоновой кислоты и карбонильные соединения на основе кислоты Мельдрума. С целью препаративного синтеза β-трикетонов с различными карбонильными акцепторами, изучения их строения и химических превращений нами получены этиловые эфиры 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой кислоты (1) и 2-гидрокси-2-(3-гидрокси-4-метил-2,5-диоксо-3-циклопентенилиден)уксусной кислоты (2) конденсацией Клайзена ацетона или 2-бутанона (метилэтилкетона) с диэтилоксалатом в присутствии натрия при кипячении смеси в бензоле:
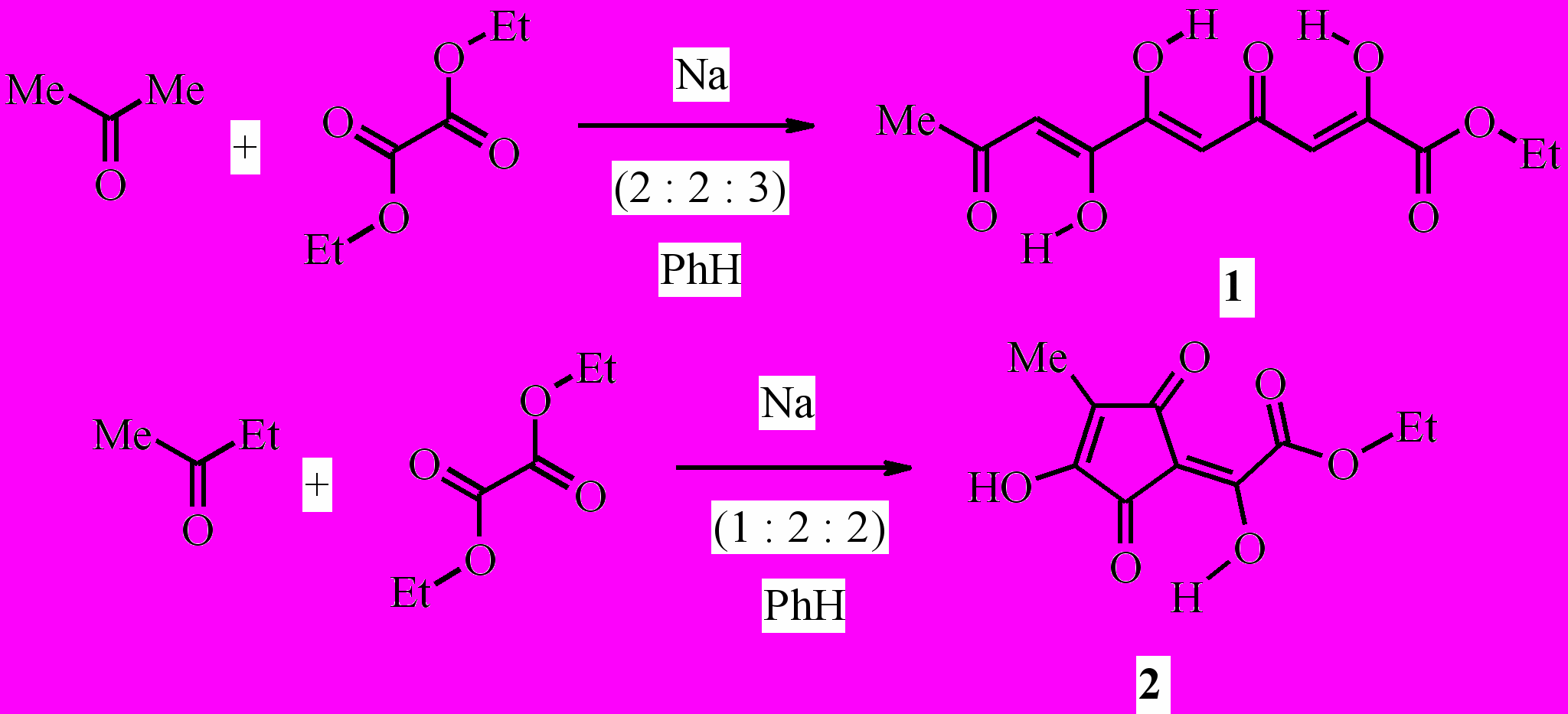
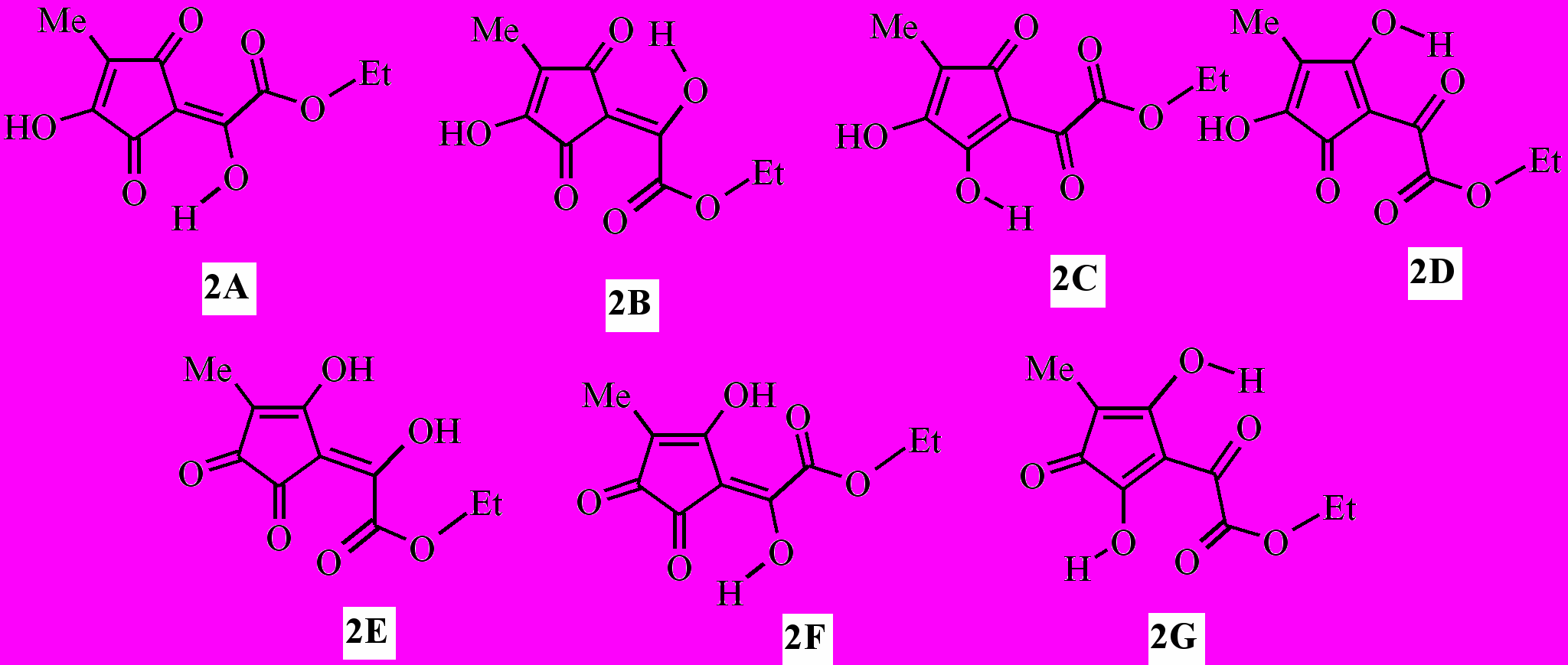
В твёрдом состоянии эфир (2) имеет две высокочастотные не связанные водородной связью карбонильные группы и OH-хелатный цикл со связанным ВМВС карбонилом. Эти данные не противоречат наиболее вероятным формам (2A), (2B), (2C) и (2D) среди семи возможных таутомеров (2A) – (2G). Что касается остальных форм, то в спектре структуры (2E) должны были бы наблюдаться полосы трёх карбонильных групп с частотой не менее 1680 см-1, а в спектрах форм (2F) и (2G) – только одного не связанного водородной связью карбонила цикла, но в действительности рисунок спектра иной. Данные спектроскопии ЯМР 1H и масс-спектрометрии, к сожалению, не позволяют однозначно установить прототропную форму эфира (2) в растворе среди указанных таутомеров. Однако формы (2E), (2F) и (2G) гораздо менее вероятны, так как сигналы гидроксильных групп в спектре ЯМР 1H (CDCl3) неравноценны и значительно отдалены друг от друга – на 2.35 м.д., только одна из этих OH-групп связана ВМВС в хелатный цикл. Из четырёх наиболее вероятных таутомерных форм (2A)–(2D) две первые представляют собой геометрические изомеры (Z- и E-), две последние – региоизомеры, а формы (2C) и (2D) очень близки и, возможно, практически не отличимы от соответствующих им прототропных форм (2A) и (2B).
По совокупности данных сделан вывод о следующем возможном резонасно стабилизированном строении эфира (2): имеются OH-хелатный цикл, сопряжённый с кольчатым β-дикарбонильным звеном, и частично делокализованная кратная связь кольца:
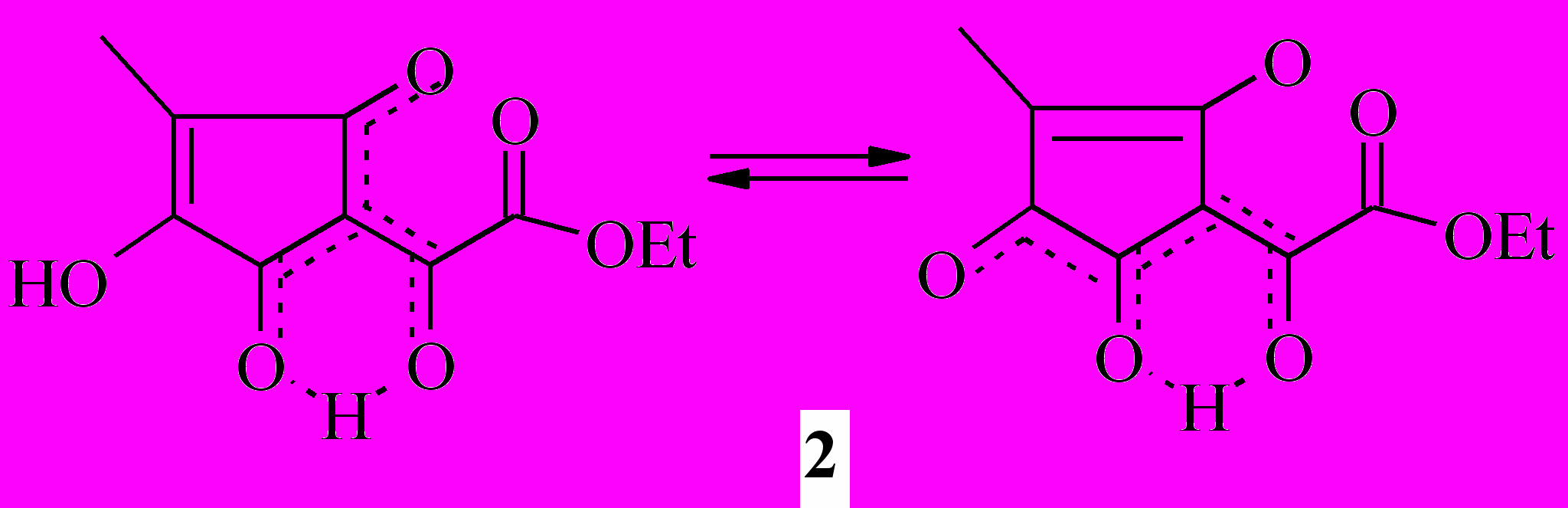
Степень туннельной миграции протона в хелатных циклах различная, и это обуславливает появление в спектрах второй (кроме сложноэфирной) карбонильной группы и одного не связанного водородной связью гидроксила.
Нами изучены химические превращения поликетидов на основе бис- и полиокса-1,3-диенов, которые будут обсуждаться более подробно ниже. Здесь мы приводим лишь некоторые данные о свойствах и реакционной способности наиболее важных в препаративном и прикладном отношении пента- и гексакарбонильных структур.
Известно, что 1,2-ди- и 1,3,4- трикарбонильные соединения легко образуют оксохиноксалиновые производные в реакциях с о-фенилендиамином. 3(2)-[Оксоалкил(иден)]производные хиноксалин-2(3)-онов широко используются в органическом синтезе и проявляют высокую биологическую активность. Нами разработан очень простой и удобный препаративный метод синтеза 3-(2-оксоалкилиден)-3,4-дигидрохиноксалин-2(1H)-онов (3a-c), а также 4,5-дигидрокси-1-[3-оксо-3,4-дигидрохиноксалин-2(1H)-илиден]-3,5-октадиен-2,7-диона (4) и 3-(2,3-дигидрокси-4-метил-5-оксо-1,3-циклопентадиен-1-ил)хиноксалин-2(1H)-она (5) реакцией ацетона, пинаколина, ацетофенона или 2-бутанона с диэтилоксалатом в присутствии натрия при кипячении смеси в бензоле с последующей обработкой уксусной кислотой и о-фенилендиамином.
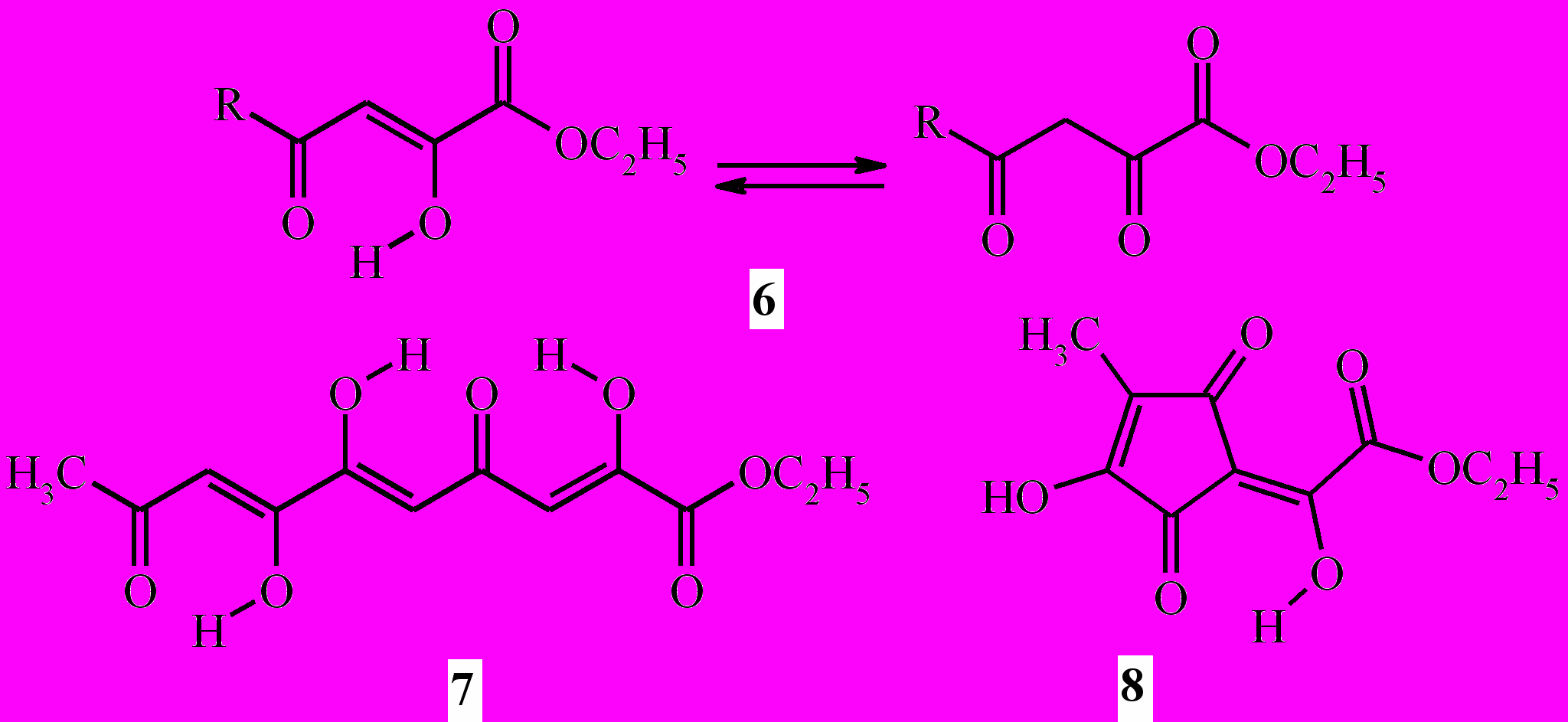
Хиноксалиноны (3a-c) и (4) образуются в результате взаимодействия промежуточных продуктов эквимолярной конденсации Клайзена метилкетонов с диэтилоксалатом – этиловых эфиров 2-гидрокси-4-оксо-2-алкеновых (ацилпировиноградных) кислот (6) и 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой кислоты (7) – с о-фенилендиамином.
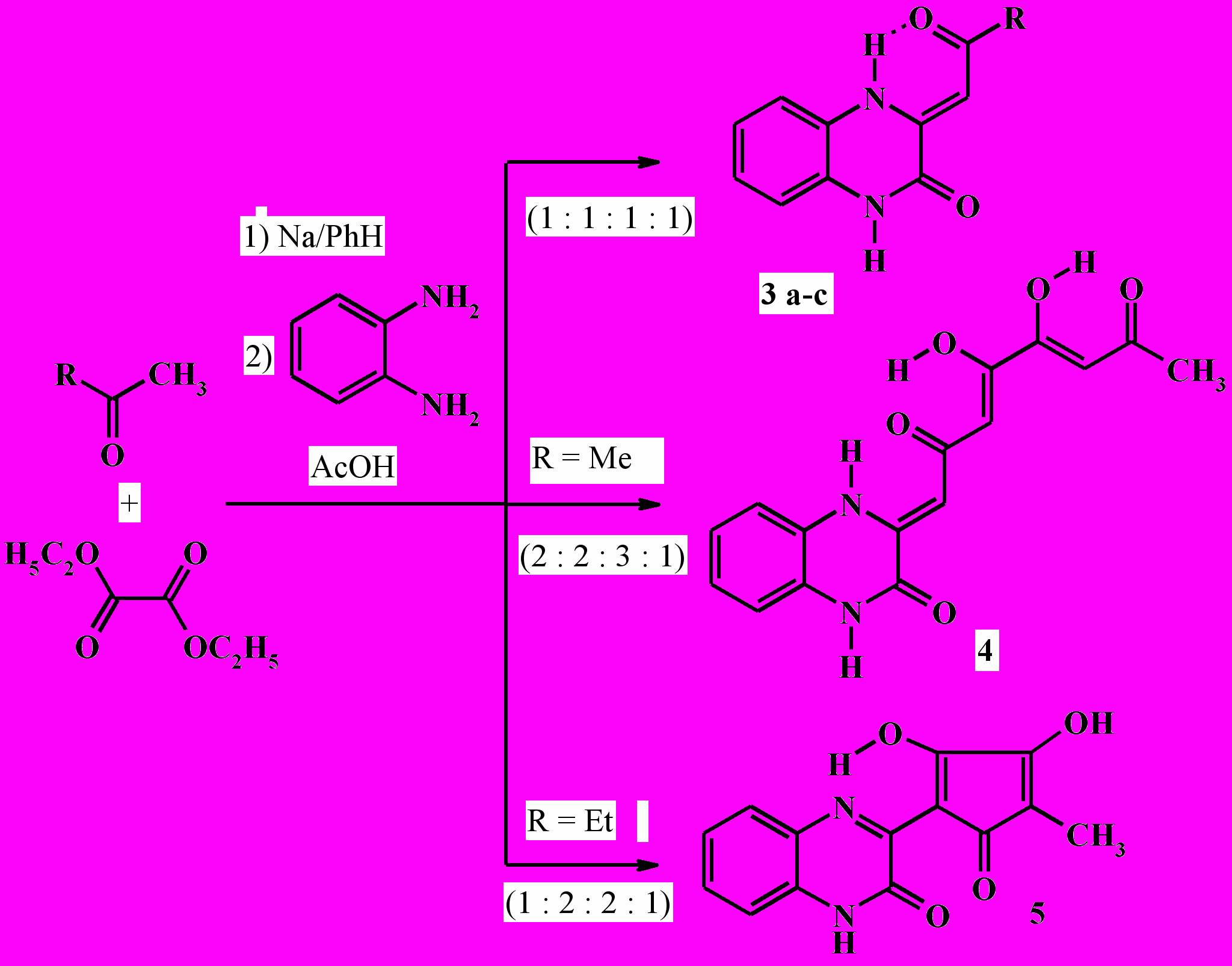
3a: R = Me; b: R = tBu; c: R = Ph
Первоначально образующийся в результате конденсации 2-бутанона с диэтилоксалатом (в соотношении 1:2) этиловый эфир 2-гидрокси(3- гидрокси-4-метил-2,5-диоксо-3- циклопентен-1-илиден)уксусной кислоты (8) также реагирует с о-фенилендиамином, образуя целевое соединение (5):
Таким образом, предложен простой и удобный препаративный метод получения 3-[оксо(цикло)алкил(иден)]замещённых хиноксалин-2(1H)-онов, который может успешно применяться в синтезе также и других разнообразных ацилметильных (оксоилиденовых) производных хиноксалина и 1,4-бензоксазина.
В отличие от пента- и гексакарбонильных соединений, часто образующих сложные смеси продуктов как в нуклеофильных, так и электрофильных реакциях, или реагирующих со значительным смолообразованием, более простые 1,3,4,6-тетракарбонильные системы гладко взаимодействуют с этими реагентами. При галогенировании 1,3-дикарбонильных соединений (ДКС) образуются 2-галогенпроизводные, проявляющие, в отличие от многих исходных ДКС, выраженное противомикробное действие. 2-Галоген-ДКС используются в синтезе антимикробных препаратов, например, теброфена. Функционализация ДКС введением как атомов галогена в β-положение, так и карбоксильной группы, приводит к биологически активным 3-галогензамещённым ацилпировиноградным кислотам, для которых получены эфиры, амиды и 2-иминопроизводные. Соединения, содержащие галоген по сравнению с неактивными или малоактивными аналогами без галогена в дикарбонильном звене обладают значительным бактериостатическим эффектом и практически не токсичны.
Бис-ДКС, имеющие в своём составе два сближенных β-дикарбонильных звена, – 1,3,4,6-тетракарбонильные соединения (1,6-диоксо-3,4-диенолы) (10, 11) как правило, не обладают существенным противомикробным действием. По аналогии с производными ацилпировиноградных кислот можно было a priori предполагать, что модификация структуры 1,3,4,6-тетраоксосоединений введением атомов галогена в дикарбонильные фрагменты их молекул приведёт к появлению заметного бактериостатического эффекта.
Нами получены 1,6-дизамещённые 2,5-дибромгексан-1,3,4,6-тетраоны ( 11a,b) и 2,2,5,5-тетрагалогенгексан-1,3,4,6-тетраоны (12a-p) при действии брома или хлора на 1,6-дизамещённые гексан-1,3,4,6-тетраоны (9a-k). Соединения 11a,b существуют в твёрдом состоянии в 3,4-диоксоформе (11A), а в растворах – в 3,4-диенольной форме (11B) или смеси таутомеров 11A и 11B. В результате бромирования (4Z)-амидов 6-арил-3,4-дигидрокси-6-оксогекса-2,4-диеновых кислот (10a-e) с препаративным выходом выделены как монобромзамещённые соединения – эфиры (2Z,4E)-2-аминокарбонил-6-арил-5-бром-3,4-дигидрокси-6-оксогекса-2,4-диеновых кислот (13a-c), так и дибромпроизводные – амиды 6-арил-2,5-дибром-3,4,6-триоксогексановых кислот (14a-d).
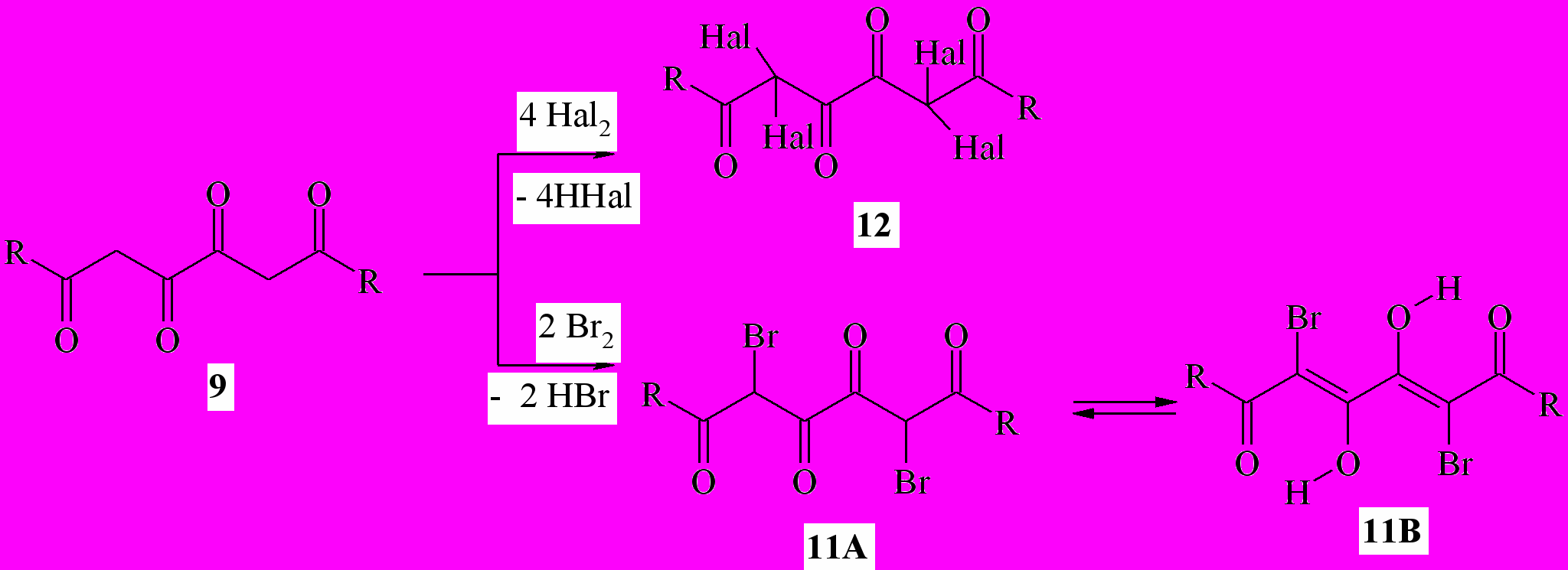
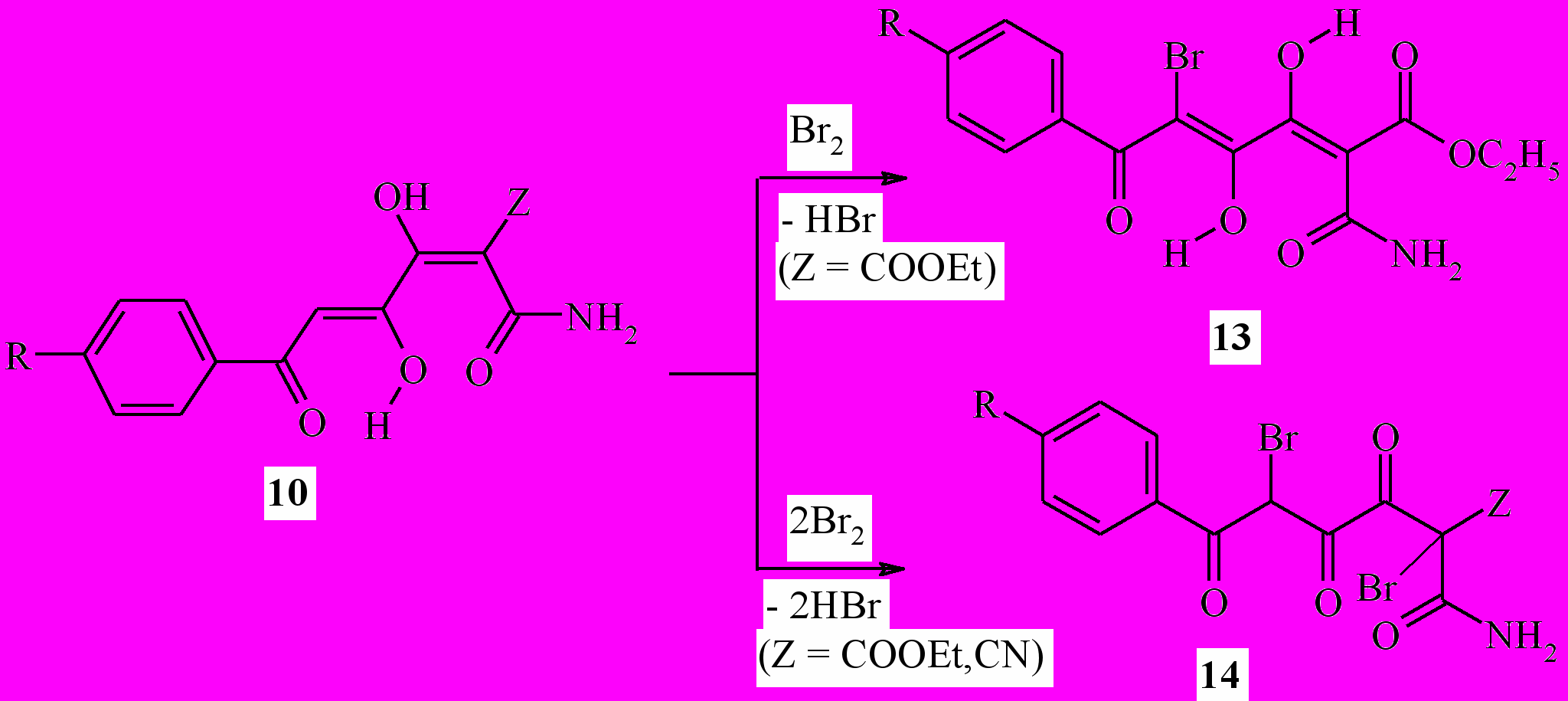
9а,11а: R=tBu; 9b,11b: R=Ph; 9c: R=4-MeC6H4; 11d: R=2,4-Me2C6H3; 11e: R=2,4,6-Me3C6H2; 11f: R=3-MeOC6H4; 11g: R=4-MeOC6H4; 11h: R=4-BrC6H4; 11i: R=4-ClC6H4; 11j: R=4-FC6H4; 11k: R=1-C10H7; 10а,14а: R=H, Z=CO2Et; 10b,10b: R=H,Z=CN; 10c,14c: R=Me,Z=CO2Et; 10d: R=Br, Z=CO2Et; 10e: R=Cl, Z=CO2Et; 12а: R=tBu,Hal=Cl; 12b: R=Ph, Hal=Br; 12c: R=Ph, Hal=Cl; 12d: R=4-MeC6H4,Hal=Cl; 12e: R=2,4-Me2C6H3, Hal=Br; 12f: R=2,4-Me2C6H3, Hal=Cl; 12g: R=2,4,6-Me3C6H2, Hal=Cl; 12h: R=3-MeOC6H4,Hal=Cl; 12i: R=4-MeOC6H4,Hal=Cl; 12j: R=4-BrC6H4, Hal=Br; 12k: R=4-ClC6H4,Hal=Br; 12l:R=4-ClC6H4,Hal=Cl; 12m: R=4-FC6H4, Hal=Br; 12n:R=4-FC6H4,Hal=Cl; 12o:R=1-C10H7,Hal=Br; 12p: R=1-C10H7, Hal=Br; 13а:R=H; 13b: R=Br; 13c: R=Cl; 14d: R=Cl, Z=CO2Et;
ъ
Известно, что ароилпировиноградные (4-арил-2-гидрокси-4-оксо-2(Z)-бутеновые) кислоты и их производные: эфиры, амиды, гидразиды успешно используются в органическом синтезе. Известные методы синтеза ароилпирувамидов обычно включают несколько стадий и являются длительными и достаточно трудоёмкими.
Благодаря наличию широкого спектра биологической активности у амидов ацилпировиноградных кислот у этих соединений были подробно изучены химические превращения с целью проследить изменение биологических свойств при модификации структуры. NH-Нуклеофильные реакции ацилпирувамидов изучены достаточно подробно, а сведения об их реакциях с CH-нуклеофилами до наших исследований отсутствовали.
Ранее были изучены реакции β-дикарбонильных соединений – ацетоуксусного и бензоилуксусного эфиров – с ацетил- и бензоилметилентрифенилфосфоранами, в результате выделены продукты C-ацилирования последних и дальнейшей гетероциклизации – 2,6-дизамещенные 4H-пиран-4-оны и 4-ацилметилен-4H-пираны. Мы установили, что ариламиды 4-арил-2-гидрокси-4-оксо-2(Z)-бутеновых (ароилпировиноградных) кислот (15а-f) легко взаимодействуют с эфирами трифенилфосфоранилиденуксусной кислоты, образуя продукты олефинирования Виттига по α-карбонильной группе – эфиры 5-арил-3-ариламинокарбонил-5-оксо-3(Z)-пентеновых кислот (17а-f) и трифенилфосфиноксид. В результате этой реакции с фениламидом 15g выделен аддукт с трифенилфосфиноксидом (18) состава 1:1. Нам не удалось разделить комплекс 18 на компоненты с помощью обычных методов. Образование такого аддукта не является неожиданным, хорошо известны устойчивые комплексы трифенилфосфиноксида с карбонильными соединениями:
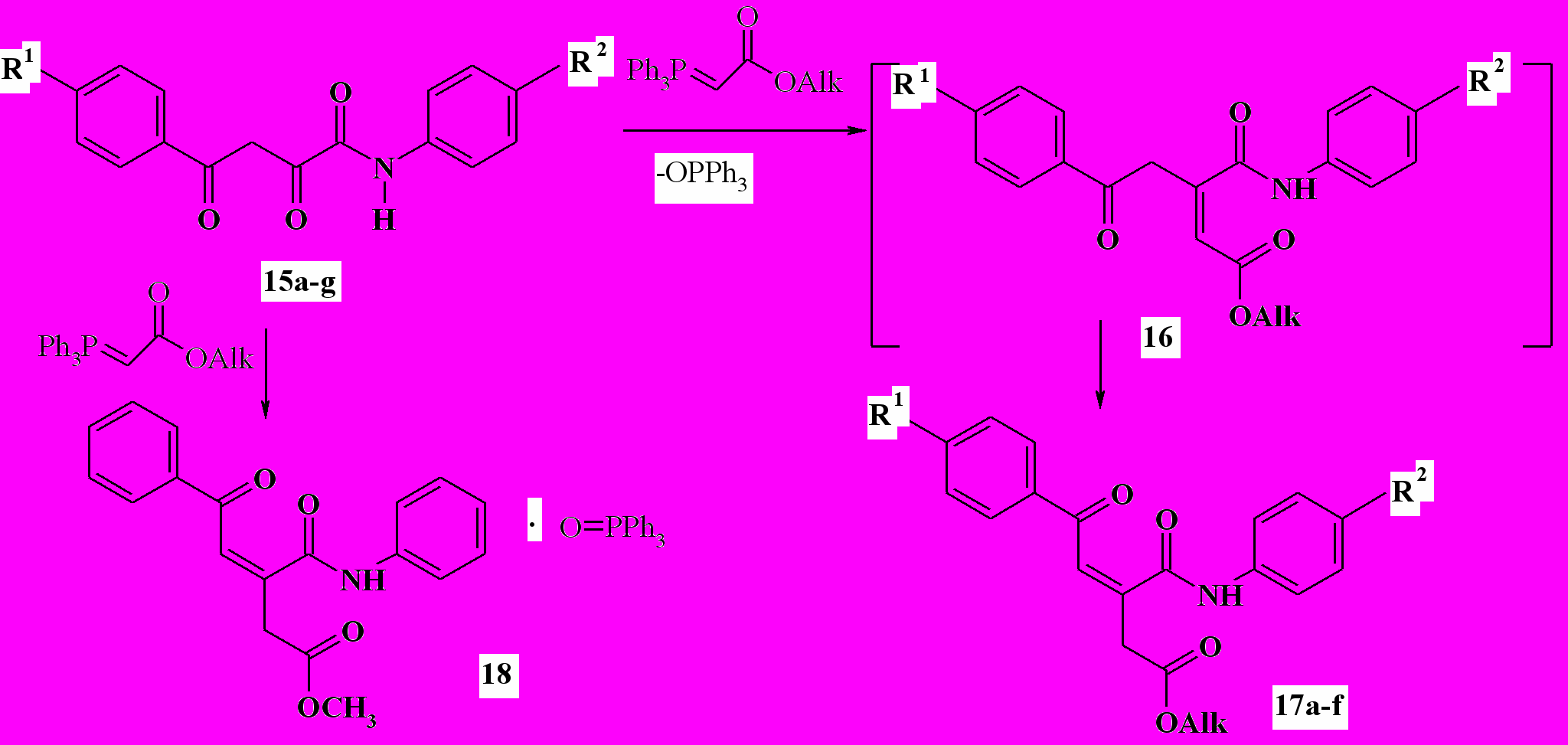
15-17a: Alk=Me,R1=H,R2=Me; b: Alk=Me,R1=H,R2=OMe; c: Alk=Me, R1=Me, R2=OMe; d: Alk=Me,R1=Br,R2=H; e: Alk=Me, R1=Cl,R2=OMe; f: Alk=Et, R1=Br, R2=Me; 15g: Alk=Me,R1=R2= H;
Ацилпировиноградные кислоты и их эфиры, в отличие от ароилпирувамидов, реагируют с фосфоранами со значительным смолообразованием, и выделить индивидуальные соединения из таких реакционных смесей нам не удавалось. Вместе с тем лактоны γ-енольной формы ароилпировиноградных кислот – 5-арилфуран-2,3-дионы – легко взаимодействуют с метиленфосфоранами, образуя с препаративными выходами различные продукты, которые обсуждаются нами далее.
Известно, что реакция Виттига 5-арилфуран-2,3-дионов, не замещённых в положении 4 цикла, а также 4-галоген- или 4-метилпроизводных, с метилентрифенилфосфоранами приводит к региоселективному моно-олефинированию по лактонной карбонильной группе.
В результате взаимодействия 4-бензоил-5-фенилфуран-2,3-диона ( 19a) с ацетилметилентрифенилфосфораном (20a) и метиловым эфиром трифенилфосфоранилиденуксусной кислоты (20b) вместо ожидаемых конечных продуктов реакции Виттига – 2- оксоилиденфуран-3(2H)-онов (21b: R = CH3, OCH3) – нами неожиданно были выделены соединения, содержащие в своем составе фосфор. По совокупности спектральных данных полученным веществам было придано строение аддуктов 4-бензоил-2-гидрокси-2-(2-оксопропил)-5-фенилфуран-3(2H)-она или метилового эфира 4-бензоил-2-гидрокси-3-оксо-5-фенил-2,3-дигидрофуран-2-илуксусной кислоты с трифенилфосфиноксидом (23a,b) состава 1:1:
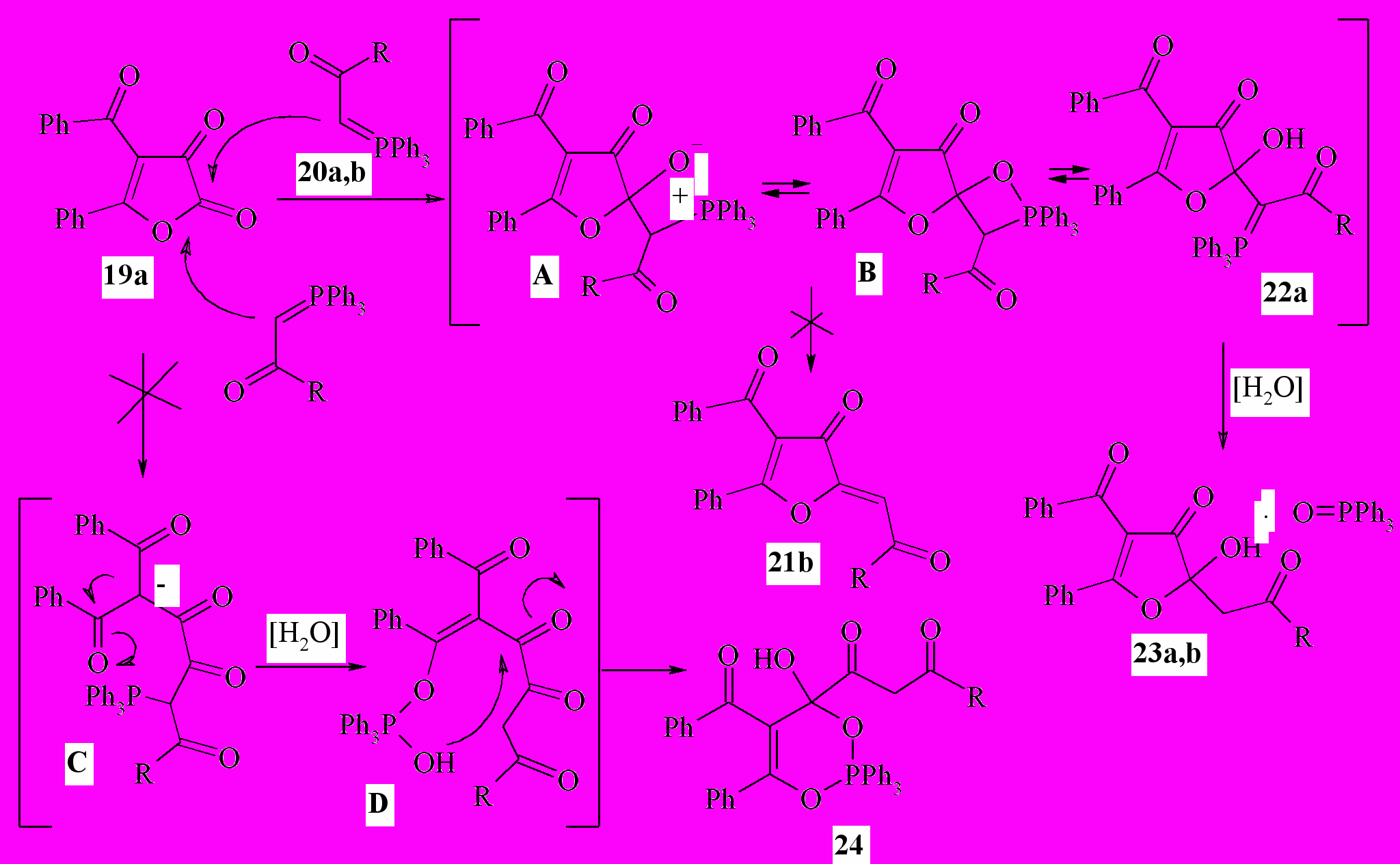
20,23a: R=Me; b: R=OMe;
Реакция фуран-2,3-диона 19 с илидами 20a,b не может протекать по альтернативной схеме с образованием производных 1,3,2-λ5- диоксафосфинана (24) через вероятные цепные оксо-интермедиаты С и D:
Взаимодействие 4-бензоил-5-фенилфуран-2,3-диона 19a с 20a,b первоначально осуществляется по механизму реакции Виттига через вероятный бетаиновый А или оксафосфетановый B интермедиаты с промежуточным образованием кольчатого илида 22a. Последующий его гидролиз под действием влаги воздуха приводит к отщеплению трифенилфосфиноксида и формированию комплекса с гетероциклическим звеном 23a,b. Очевидно, последний процесс протекает значительно легче, чем возможное традиционное элиминирование трифенилфосфиноксида с альтернативным образованием олефинов 21b. Менее вероятная возможность протекания конкурентной реакции с формированием диоксафосфинанового кольца 24 через промежуточные цепные оксо-илиды C и D тоже не реализуется. Отметим также, что региоселективность C-нуклеофильной атаки реагентов 22a,b по лактонному карбонилу соединения 21 подтверждает данные о большей электрофильности атома углерода группы C2=O по сравнению с кетонной группой C3=O фуран-2,3-дионов.
Известно, что поликарбонильные системы, содержащие четыре и более сближенных 1,2- и 1,3-дикарбонильных фрагментов, в растворах склонны к прототропным превращениям, имеют разнообразные цепные и кольчатые таутомерные формы и легко вступают в кольчато-кольчатые интерконверсии. Актуальным, таким образом, является разработка новых методов получения кольчатых аналогов поликетидов – производных 2-гидрокси-3-оксофурана и 4-пиранона, а также их производных и исследование структуры и химических превращений этих субстратов. 2-Гидрокси-2,3-дигидрофуран-3-оны успешно используются в органическом синтезе. Доступные методы синтеза 5-арил-2-гидроксифуран-3(2H)-онов обычно включают несколько стадий и не являются препаративными. Нами разработан очень простой и удобный одностадийный способ получения эфиров 2-гидрокси-3-оксо-2,3-дигидрофуран-2-илуксусной кислоты (25a,b) реакцией этилацетата с диэтилоксалатом и метилкетонами в присутствии гидрида натрия с последующей обработкой смеси соляной кислотой:
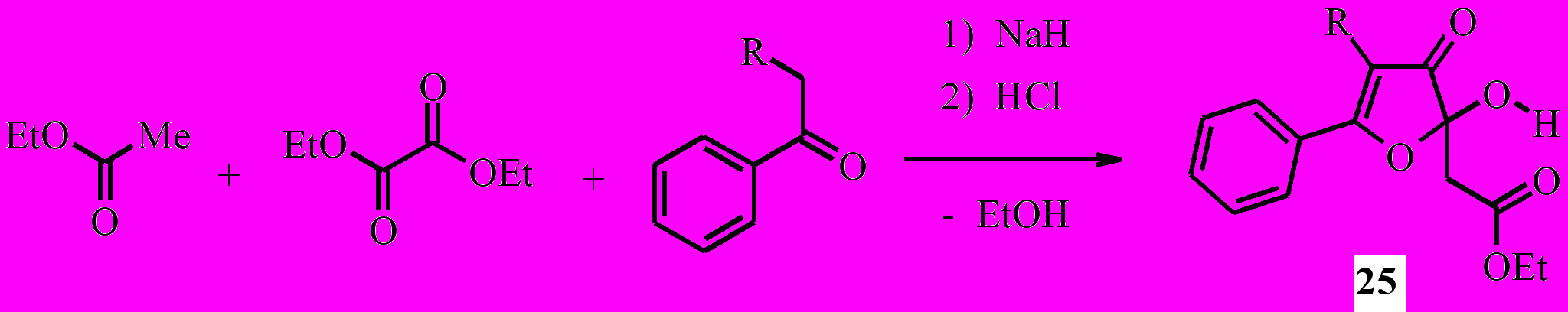
25a: R=H; b: R=PhCO
Кроме соединения 25a из реакционной смеси этилацетата с диэтилоксалатом и ацетофеноном с выходом 27% выделен побочный продукт – Z-2-гидрокси-4-фенил-4-оксо-2-бутеновая (бензоилпировиноградная) кислота, идентифицированная сравнением с известным образцом.
2. Синтез и некоторые реакции моно- и полиядерных азагетероциклов на основе (O,N)гетеро-1,3-диенов и их производных
В предыдущем разделе мы рассмотрели методы получения, особенности строения и некоторые свойства поликарбонильных систем и их кольчатых производных на основе активированных акцепторами окса-1,3-диенов – синтетических O-эквивалентов гетеро-1,3-диенов. В этом разделе мы переходим к обсуждению химии разнообразных азагетероциклов как кольчатых N-аналогов последних. Заметим, что представленная в таком разрезе системная характеристика ненасыщенных O- и N-гетероциклов, состоящих из фрагментов гетеро-1,3-диенов и имеющих эти звенья в боковых цепях, в отечественной литературе рассматривается впервые. Ранее было показано структурное и реакционное химическое разнообразие 1,2,4-трикарбонильных систем на примере ацилпировиноградных кислот и их ближайших кольчатых O-производных. Обсуждение результатов нашей работы в настоящей главе мы начнём с синтеза моноядерных диазолов и диазинов, а затем представим материал по полиядерным моно- и полиазагетероциклам – производным индола, хинолина, некоторым гетероаннелированным структурам (карболинам, индолохиназолинам, пиридазинохиназолинам и др.).
Известно, что (гет)ароилпировиноградные кислоты, их эфиры и амиды реагируют с гидразинами с образованием производных 5-(гет)арил-1H-пиразол-3-карбоновых кислот. Нами в результате взаимодействия пивалоилпировиноградной (2-гидрокси-5,5-диметил-4-оксо-2-гексеновой) кислоты (26) с гидразидами ароматических карбоновых кислот в мягких условиях с препаративным выходом получены 2-ароилгидразоно-5,5-диметил-4-оксогексановые кислоты (27a,b, форма A), в растворах которых присутствует минорный кольчатый пиразолиновый таутомер – 1-ароил-5-трет-бутил-5-гидрокси-4,5-дигидро-1H-пиразол-3-карбоновые кислоты (форма B). Строение последних хорошо согласуется с таковым полученных ранее амидов 5-арил-5-гидрокси-2-пиразолин-3-карбоновых кислот
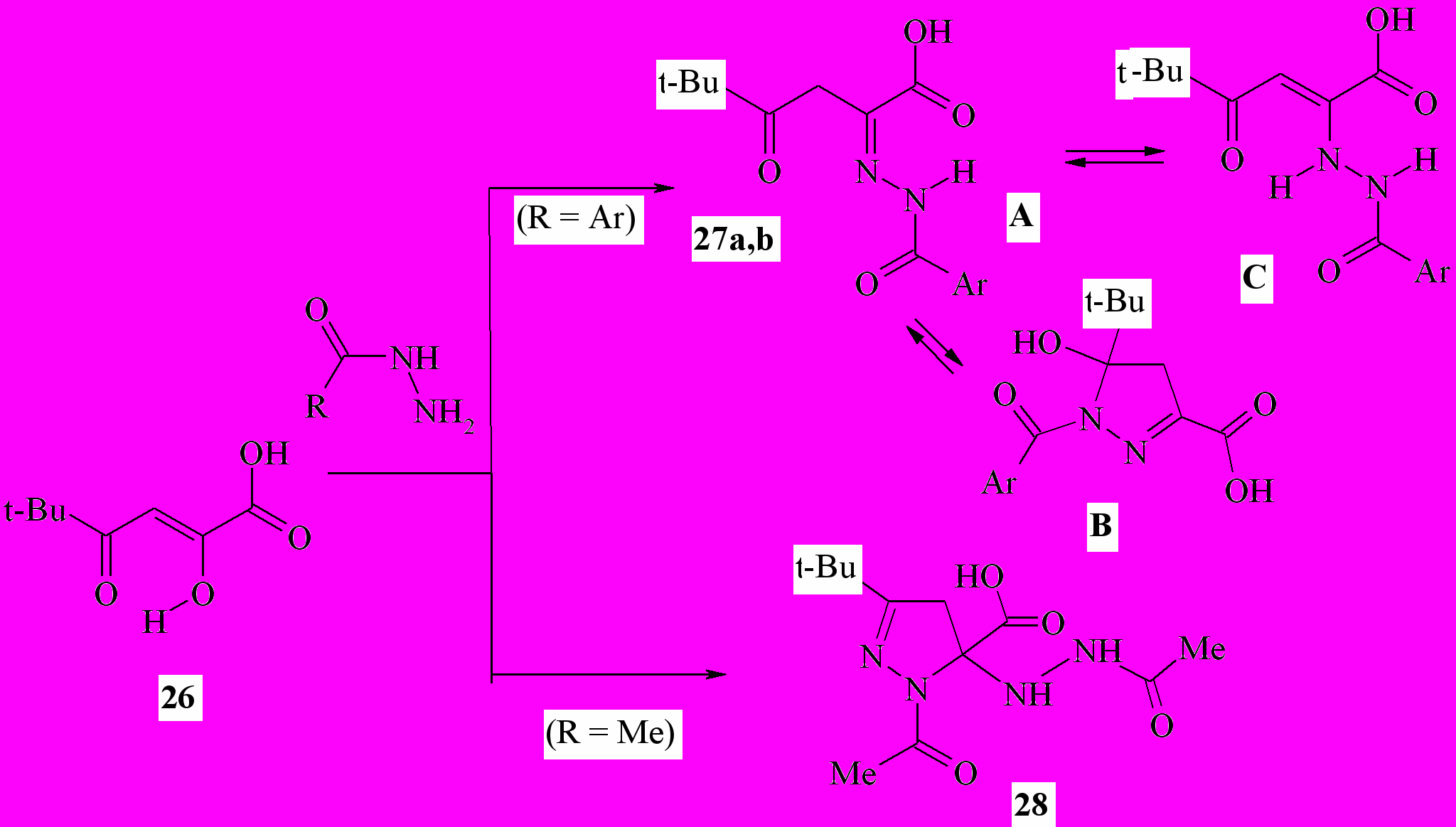
27a: Ar = 4-MeC6H4; b: Ar = 4-MeOC6H4;
Кроме равновесных структур A и B в растворах соединений 27a,b отмечено небольшое количество (до 7%) цепной енгидразино-формы 2-(2-ароилгидразино)-5,5-диметил-4-оксо-2-гексеновых кислот (C). Отметим, что соединения 27a,b в кристаллах представлены только NH-хелатной формой C, стабилизированной ВМВС типа >N-H …O=C< . При действии ацетилгидразина на кислоту 26 нами неожиданно был выделен ранее не известный устойчивый кольчатый продукт присоединения двух молекул реагента по α- и γ-карбонильным группам субстрата – 1-ацетил-5-(2-ацетилгидразино)-3-трет-бутил-4,5-дигидро-1H-пиразол-5-карбоновая кислота (28).
Одними из близких кольчатых производных ацилпировиноградных кислот являются производные 3-оксофурана: лактоны γ-енольной формы указанных кислот – фуран-2,3-дионы и 2-илиденпроизводные последних. Ранее было установлено, что реакции 2-илиденфуран-3(2H)-онов с гидразином, арил- или гетарилгидразинами весьма разнообразны и приводят к образованию гидразинопроизводных 3-оксофурана, а также циклических азолов или азинов – 3-ацилпиразолов, 3-(3-оксо-5-пиразолил)пиразолов или замещённых пиридазин-4(1H)-онов. Нами изучены особенности протекания реакции 2-оксоилиденфуран-3(2H)-онов с гидразидами карбоновых кислот и их производными по сравнению с более простыми нефункционализованными гидразинами.
В результате взаимодействия эфиров 5-арил-3-оксофуран-2(3 H)-илиденуксусных кислот (29a,b) или 5-фенил-2-[2-(4-хлорфенил)-2- оксоэтилиден]фуран-3(2H)-она (29c) с гидразидами уксусной, бензойной, анисовой, п-нитробензойной и салициловой кислот в среде этанола при кипячении с препаративными выходами (44-86%) получены продукты реакции, которые на основании спектральных данных и сравнения с известными 4-оксопиридазинами идентифицированы как 3-замещённые 6-арил-2-ацил-3-гидрокси-2,3-дигидропиридазин-4(1H)-оны (30a-h). В растворах соединений 30 присутствуют две таутомерные формы А и Б.
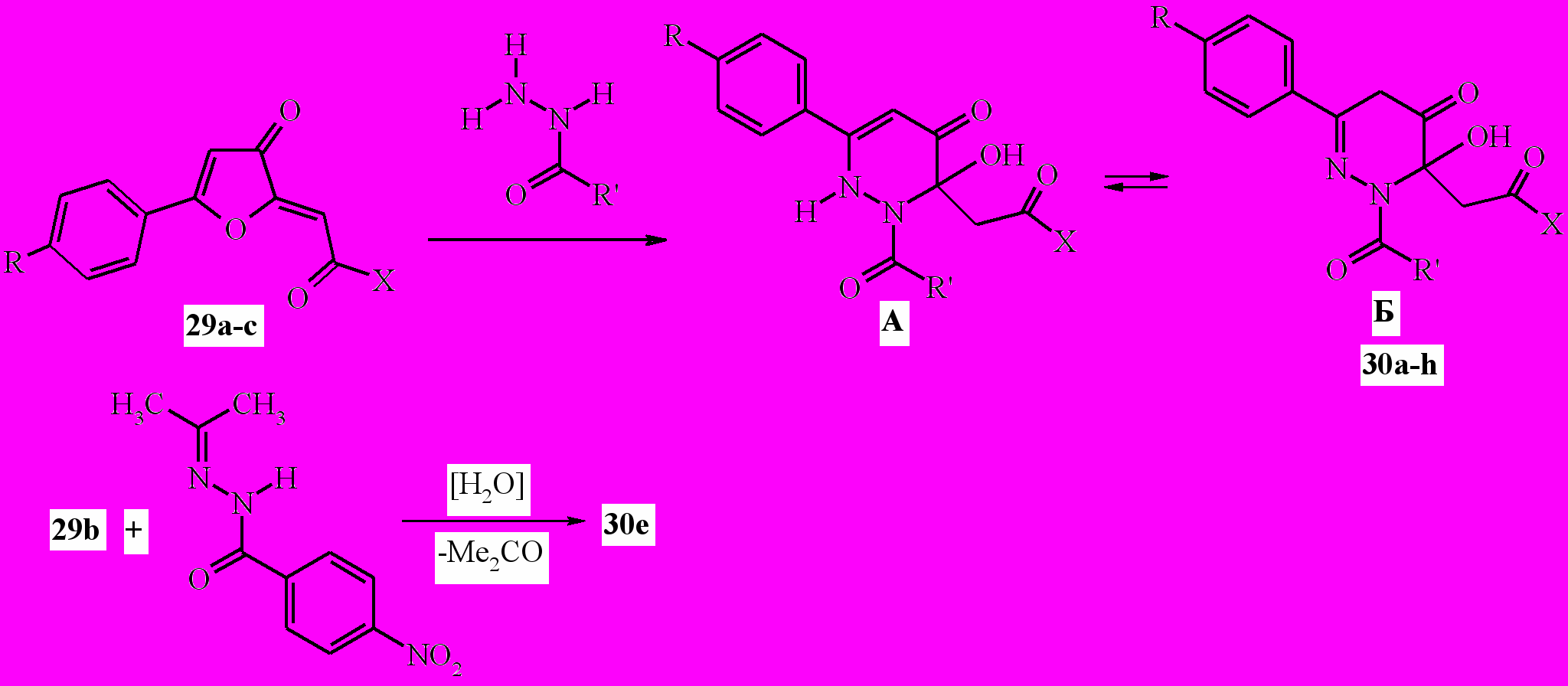
29a: X =OMe, R=Me; b: X =OMe, R=H; c: X=4-ClC6H4, R=H; 30a: X= OMe, R=R'=Me; b: X=OMe, R=Me, R'=Ph; c: X=OMe, R=H, R'=Ph; d: X = OMe, R=H, R’=2-HOC6H4; e: X=OMe, R=H,4-NO2C6H4; f: X = 4-ClC6H4, R=H, R'=Ph; g: X=OMe, R=H, R’=4-CH3OC6H4; h: X=OMe, R=H, R’=2-HOC6H4.
Пиридазинон 30e также получен при действии изопропилиден-гидразида п-нитробензойной кислоты на эфир кислоты 29b в аналогичных условиях. Следует отметить, что другие N'- илидензамещённые гидразиды, например бензилиденгидразид п-нитробензойной кислоты, в этих условиях не удалось вовлечь в реакцию с илиденфураноном 29b.
Совершенно иное направление наблюдается для реакций 2-замещённых 5-арил-3(2 H)-фуранонов с гидразидом антраниловой кислоты, и это отличие выражается в создании неизвестных ранее 2-арил-4a,5-дигидро-1H-пиридазино[6,1-b]хиназолин-4,10-дионов.
Известны способы построения практически гетероаннелированных систем на основе хиназолина: пирроло[1,2-a]хиназолинов, имидазо[2,1-b] хиназолинов, индоло[2,1-b]-хиназолинов и пиридазино[6,1-b] хиназолинов. С целью дальнейшей разработки удобных методов получения, исследования строения, химических свойств и биологической активности диоксопроизводных хиназолиновых систем нами изучено взаимодействие циклических карбонильных субстратов – 5-арил-2-ацилметилен-3(2H)- фуранонов (29) и метиловых эфиров 2-(5-арил-2-гидрокси-3-оксо-2,3-дигидро-2-фуранил)уксусной кислоты (25) – с гидразидом антраниловой кислоты. В результате реакции фуранонов 29 или 25 с этим гидразидом, протекающей при кратковременном нагревании смеси в этаноле, выделены неизвестные ранее 2-арил-4a,5-дигидро-1H-пиридазино [6,1-b]хиназолин-4,10-дионы (31a-f) с выходами 40-67%.
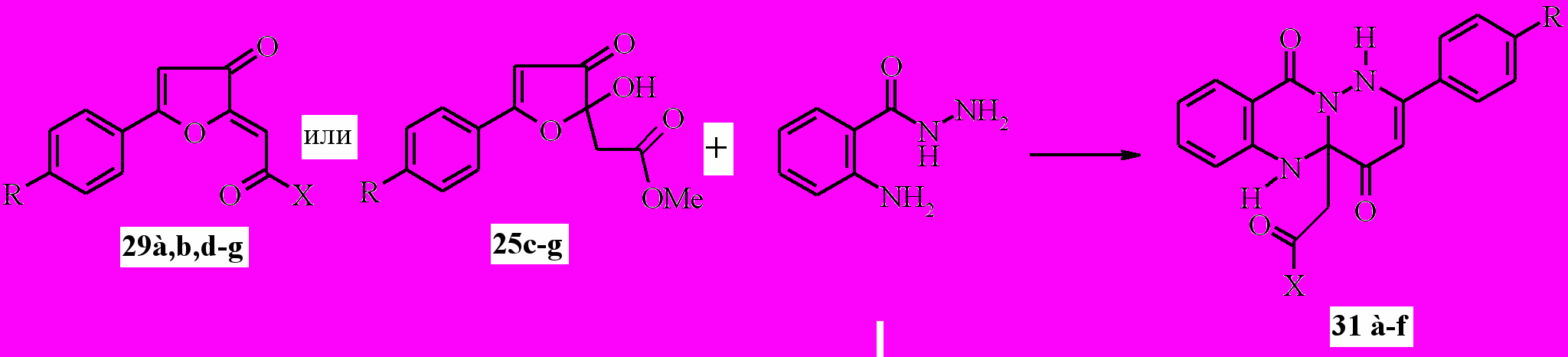
31a: X =OMe, R=Me; b: X =OMe, R=H; c: R=X =OMe; d: X=OMe, R= Br; e: X=OMe, R=Cl; f: X=4-BrC6H4, R=H;
Одним из наиболее важных и интересных в препаративном отношении, а также реакционноспособных кольчатых синтетических N- эквивалентов активированных гетеро-1,3-диенов являются оксопроизводные индола.
Известно, что индол-2,3-дионы (изатины) легко взаимодействуют с метилентрифенилфосфоранами с образованием практически значимых 3-метилен-1,3-дигидро-2H-индол-2-онов, среди которых найдены соединения, обладающие противомикробным и противосудорожным действием. С целью дальнейшего исследования химических и биологических свойств ацилметиленоксиндолов нами получены некоторыеэфиры (2Z)-(2-оксо-1,2-дигидро-3H-индол-3-илиден) уксусной кислоты (33a-l) реакцией Виттига изатинов 33a-g с алкоксикарбонилметилен-трифенилфосфоранами в бензоле или диоксане:
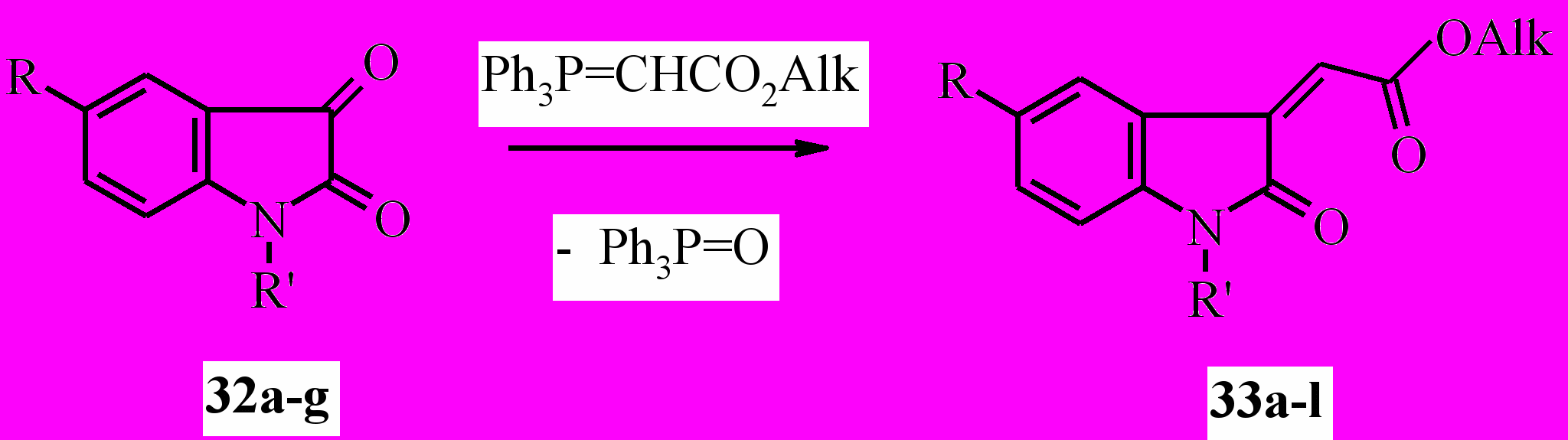
33a: R=R’=H, Alk=Me; b: R=R’=H, Alk=Et; c: R=Br, R’=H, Alk=Me; d: R=Br, R’=H, Alk=Et; e: R=NO2, R’=H, Alk =Me; f: R=NO2, R’=H, Alk = Et; g: R=H, R’=COMe, Alk=Me; h: R=H, R’=COMe, Alk = Et; i: R=Br, R’=COMe, Alk=Me; j: R=Br, R’=COMe, Alk=Et; k: R=NO2, R’=COMe, Alk =Me; l: R=H, R’=COCF3, Alk = Me;
При проведении реакции 32e с этоксикарбонилметилентрифенилфосфораном кроме обычного продукта реакции Виттига 33j нами неожиданно был выделен диэтиловый эфир 2,1'-диацетил-5,5'-дибром-1,2'-диоксо-1,1',2,2',7,8,8a-гептагидроспиро[бензо[cd]индол-6,3'-индол]-7,8-дикарбоновой кислоты (34):
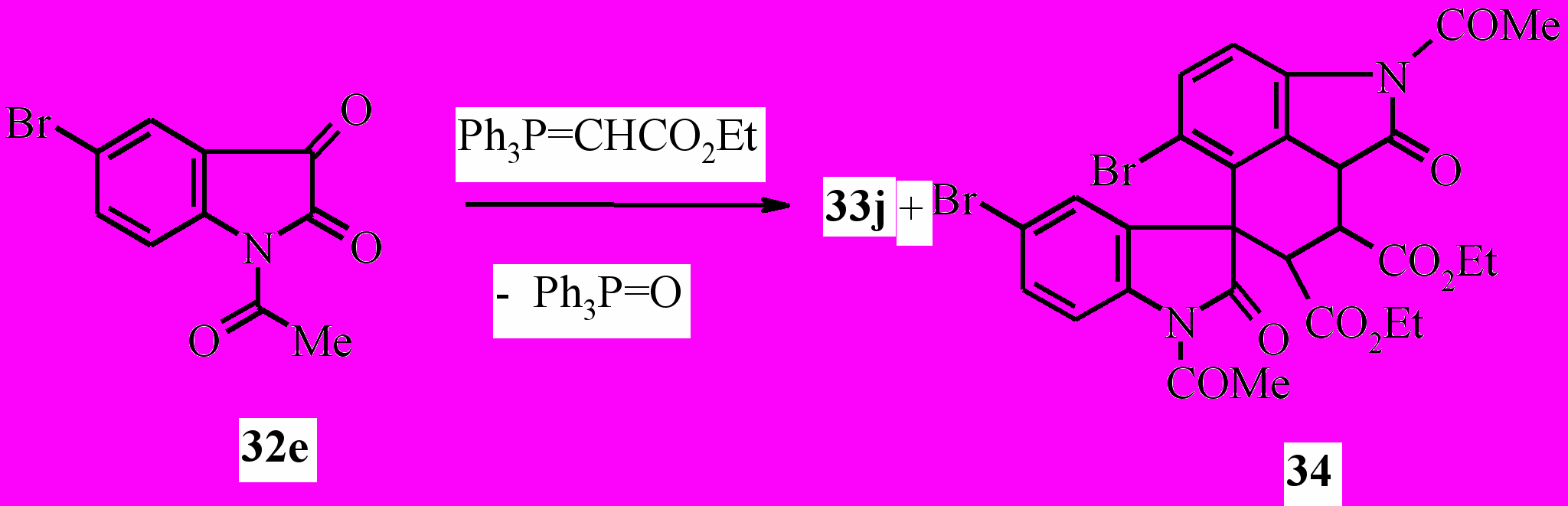
Химическая модификация структуры илиденоксиндолов приводит к разнообразным индолилпроизводным, сведения о которых мы приводим ниже.
Нами установлено, что действие ариламинов на близкие соединениям 35 по строению эфиры (2-оксо-1,2-дигидро-3H-индол-3-илиден)уксусной кислоты (33a-e) при кипячении смеси в этаноле приводит к образованию продукта региоселективного присоединения аминов по 3-экзоэтиленовой связи не в β-, как ожидалось, а в α- положение по отношению к сложноэфирной группе – эфиров 2- ариламино-2-(2-оксо-2,3-дигидро-1H-индол-3-ил)уксусных кислот (36a-h):
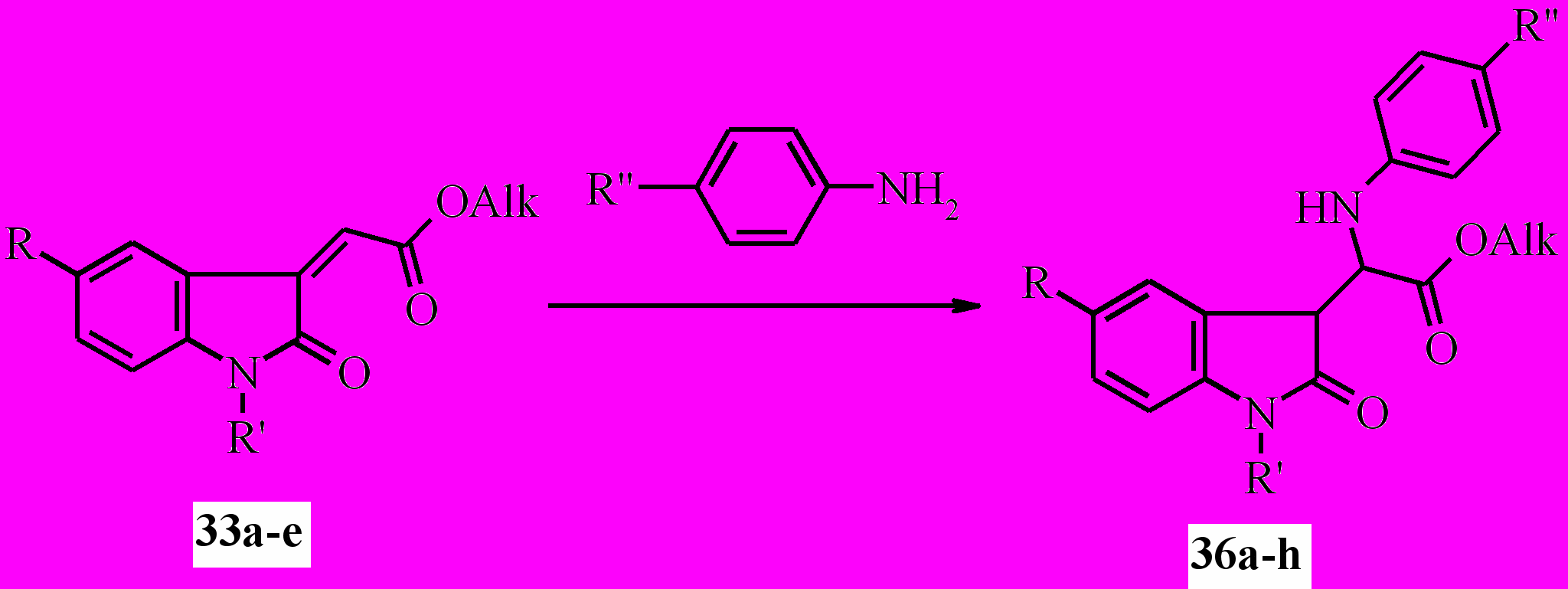
36a: R=R'=H, Alk=Me, R''=Me; b: R=R'=H, Alk=Me, R’’=OMe; c: R=R'=H, Alk = Et, R''=Me; d: R=R'=H, Alk=Et, R''=OMe; e: R=H, R'= COMe, Alk=Me, R''=Me; f: R=H, R'=COMe, Alk=Me, R''=OMe; g: R= Br, R'=COMe, Alk=Et, R''=Me h: R=NO2, R'=H, Alk=Et, R''=Me;
Илиденоксиндолы легко реагируют также и с NH-динуклеофилами, например, гидразинами, образуя как производные по илиденовому фрагменту, так и гетероаннелированные индолы.
В отличие от X-ацильных производных илиденоксиндолов близкие к ним по строению эфиры 2-(2-оксо-1,2-дигидро-3H-индол-3-илиден)уксус-ных кислот (33, X = OAlk) иначе реагируют с гидразином. В среде уксусной кислоты образуется смесь продуктов α-присоединения по экзоэтиленовой связи по отношению к сложноэфирной группе (с последующей рециклизацией) – 3,3a,5,9b-тетрагидро-1H-пиразоло[3,4-c]хинолин-1,4(2H)-дион (37) и β-присоединения – 1-ацетил-5'H-спиро[индол-3,3'-пиразолидин]-2,5'(1H)-дион (38):
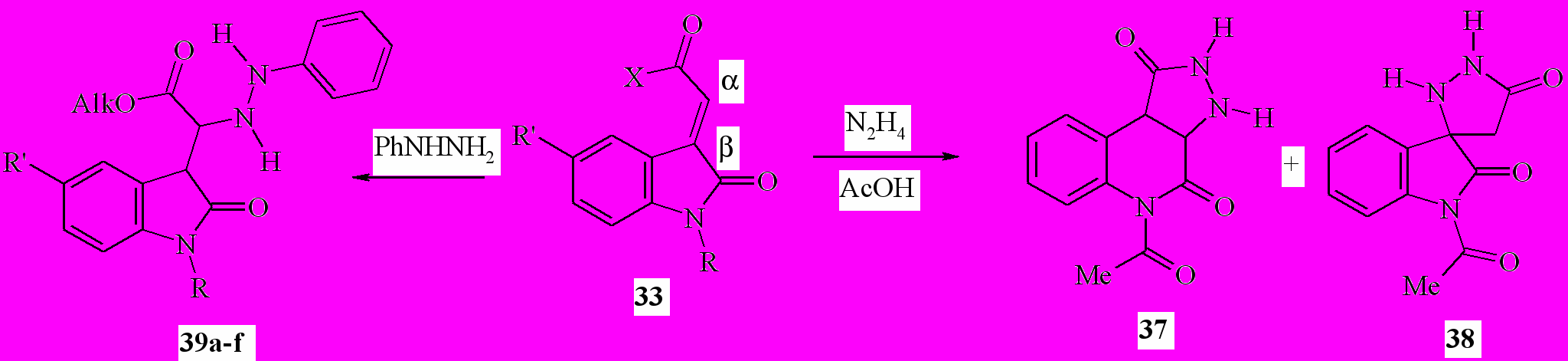
39a: Alk=Me, R=H, R'=H; b: Alk=Me, R=H, R'=Br; c: Alk=Me, R=MeCO, R' = H; d: Alk= Et, R = H, R' = H; e: Alk=Et, R=H, R'=Br; f: Alk=Et, R=MeCO, R'=H;
Мы установили, что действие фенилгидразина на эфиры 2-(2-оксо-1,2-дигидро-3 H-индол-3-илиден)уксусных кислот 33 при кипячении смеси в этаноле приводит с препаративным выходом к образованию эфиров (2-оксо-2,3-дигидро-1H-индол-3-ил)(2-фенилгидразино)уксусных кислот (39a-f). По совокупности данных, атаке гидразинами (и аминами) подвергаются четыре электрофильных центра илиденоксиндолов: при атомах C2 (NC2=O), C3 (β-положение 3-экзоэтиленовой связи), C1' (α- положение) и C2' (XC2'=O); a priori предсказать направление нуклеофильной атаки затруднительно.
Известно, что алкалоид куропитин А (40), содержащийся в тропических растениях рода Couropita, обладает высокой противогрибковой активностью. Ранее был предложен удобный препаративный метод получения куропитина реакцией изатина с хлорокисью фосфора. В продолжение начатых ранее исследований гетероаннелированных диоксо-производных хиназолина и для сравнительных биологических испытаний нами получен сам куропитин А (63) и неизвестное ранее 3,10-дибромпроизводное 41:
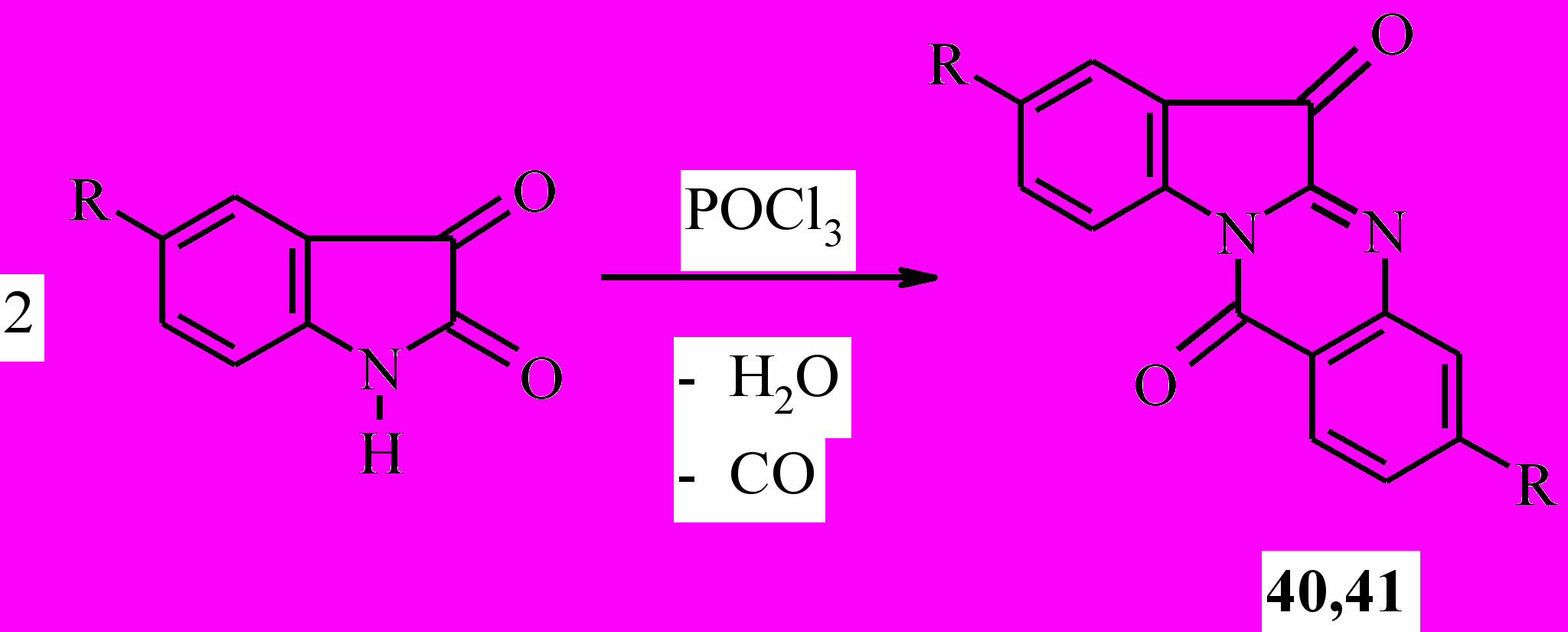
40: R=H; 41: R=Br;
3. Синтез и некоторые свойства бис-гетероциклов – синтетических эквивалентов N-гетеро-1,3-диенов
В четвёртом разделе мы переходим к обсуждению химии разнообразных бис-азагетероциклов, соединённых между собой одинарной связью. Отметим, что синтез изученных нами молекул специфичен, а их свойства резко отличаются от конденсированных (аннелированных) структур.
Синтез бис гетероциклов из производных 2-(2-оксо-1,2-дигидро-3H-индол-3-илиден)уксусных кислот
В продолжение исследований оксиндольных производных (см. раздел 3) мы приводим информацию о бис-азагетероциклических системах, содержащих ядро индола. Разнообразные 3-ацилметил(иден)производные оксиндола, как известно, обладают значительной противомикробной, противоопухолевой и противосудорожной активностью. Таким образом, поиск биологически активных соединений среди оксиндолов, связанных в положении 3 с фрагментами азотистых гетероциклов, является актуальным.
По нашим данным, атака как моно-NH-, так и SH,NH-, и NH,NH-бинуклеофилов направлена на электрофильный центр α-C2 оксиндолилиденацетатов. Мы установили, что действие о-фенилендиамина или о-аминотиофенола на эфиры 33 при кипячении смеси в этаноле или уксусной кислоте приводит с препаративным выходом к образованию 3-(2-оксо-2,3-дигидро-1H-индол-3-ил)-3,4- дигидрохиноксалин-2(1H)-онов (42a-d) или 2-(2-оксо-2,3-дигидро-1H-индол-3-ил)-2H-1,4-бензотиазин-3(4H)-онов (43a-d):
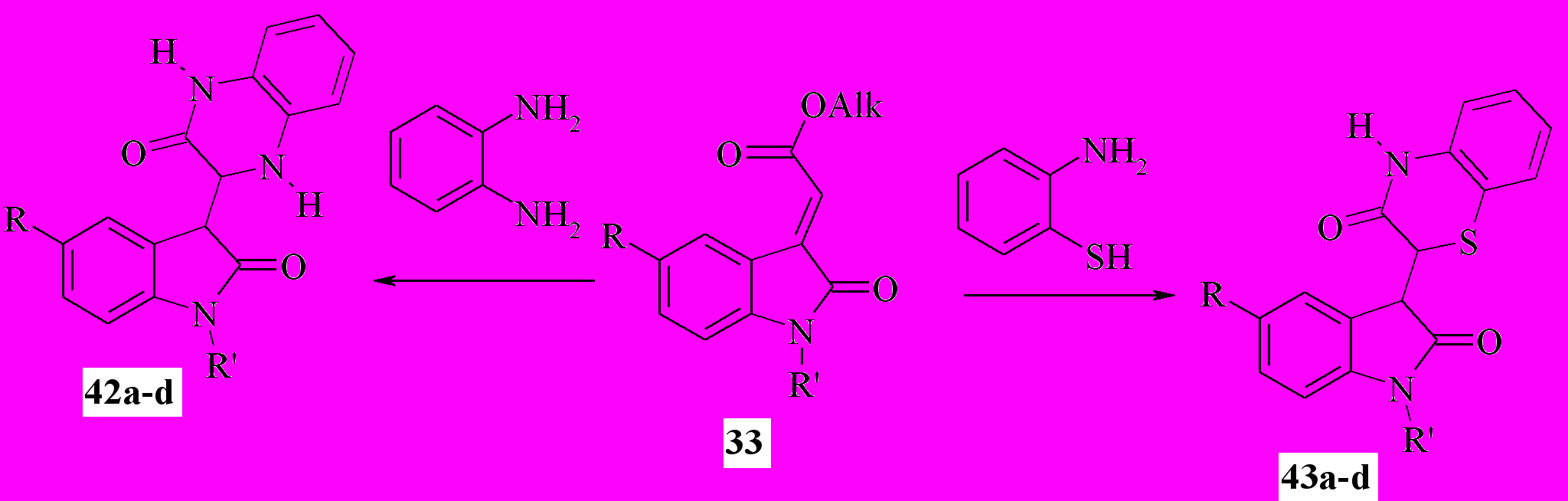
42,43a: R=R’=H; b: R=H, R’=COMe; c: R=Br, R’=H; d: R = Br, R’=COMe;
Мы также установили, что действие циклоалифатических или гетероциклических 1,2-диаминов (например, 1,2- диаминоциклогексана или 2,3-диаминопиридина) на эфиры 2-(2-оксо-1,2-дигидро-3H-индол-3-илиден) уксусной кислоты (56) при кипячении смеси в этаноле приводит с препаративным выходом к образованию 2-оксо-2,3-дигидро-1H-индол-3- илпроизводных октагидрохиноксалин-2(1H)-она (44) или, соответственно, 1,4-дигидропиридо[2,3-b]пиразин-3(2H)-она (45):
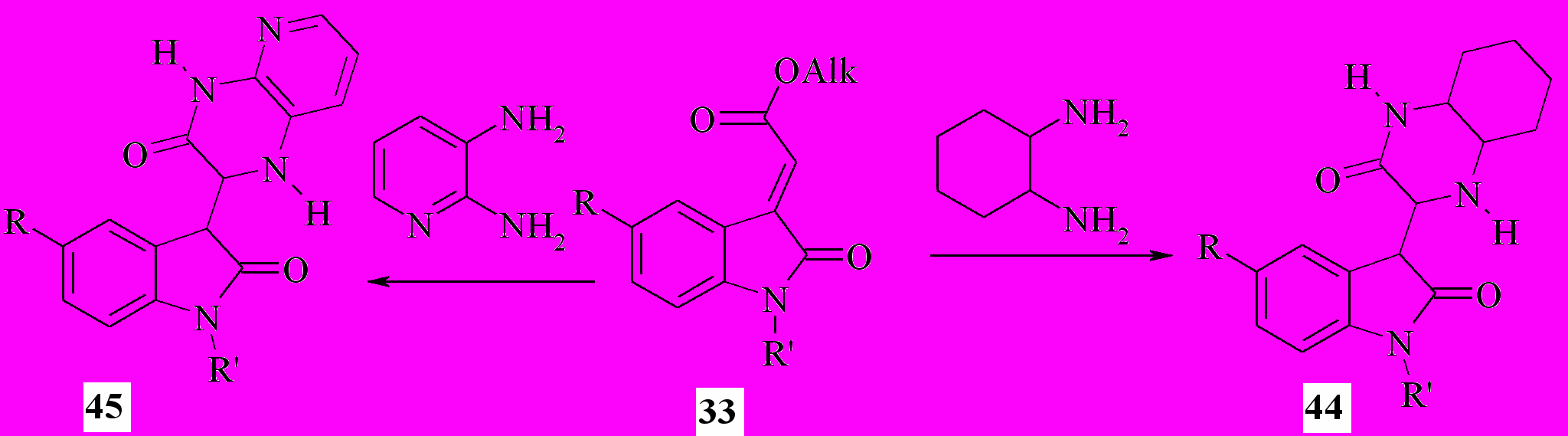
Соединения 42-45 являются продуктами региоселективного присоединения амино- или тиольной группы реагентов по экзоэтиленовой связи субстрата 42 не в β-C3'-, как можно было ожидать, а в α-C2-положение к сложноэфирному звену с последующей гетероциклизацией при участии последнего и свободной орто-аминогруппы.
Реакция оксиндолилиденацетатов с о-диаминами, по-видимому, первоначально протекает как региоселективное присоединение амино- или тиольной группы по экзоэтиленовой связи в α-положение к сложноэфирному звену. Изменение направления присоединения нуклеофилов (в α-C2-положение) по сравнению с известным (β-C3'-атака) для 3-ацилметилен-1H-индол-2-онов, вероятно, обусловлено большим электроноакцепторным влиянием сложноэфирной группы субстратов.
Синтез 3-гетарилхинолинов
Большинство синтезов 3-гетарилхинолинов сводятся к получению 1,3-дикарбонильных соединений или их дезоксоаналогов - -непредельных соединений. Исключения, в основном, составляют электроциклические реакции и синтезы, включающие аминирование ароматического ядра. В некоторых случаях, например, синтез хинолинов по Скраупу и родственные ему, непредельная или дикарбонильная система создается до формирования гетероциклического ядра, в других в ходе его формирования. Поэтому во многих синтезах используется методология, аналогичная получению непредельных и дикарбонильных соединений. Доминируют реакции альдольного типа, ацилирования (формилирования), алкилирования карбонильными соединениями различных субстратов, кросс-сочетание. Методы, в основном, различаются взаимным расположением функциональных групп участвующих в реакции. Используя ряд примеров синтезов 3- гетарилхинолинов, в этой части работы мы попытались проиллюстрировать эти идеи.
В качестве первого подхода к синтезу таких соединений мы использовали, основанный на синтезе хинолинов по Робинсону, несколько модифицировав этот метод. Из -(2-хинолил)-2-аминостиролов (46) были получены амиды 52, которые без очистки пускали в реакцию с POCl3 или полифосфорной кислотой (ПФК). Выход составил 67-78%:
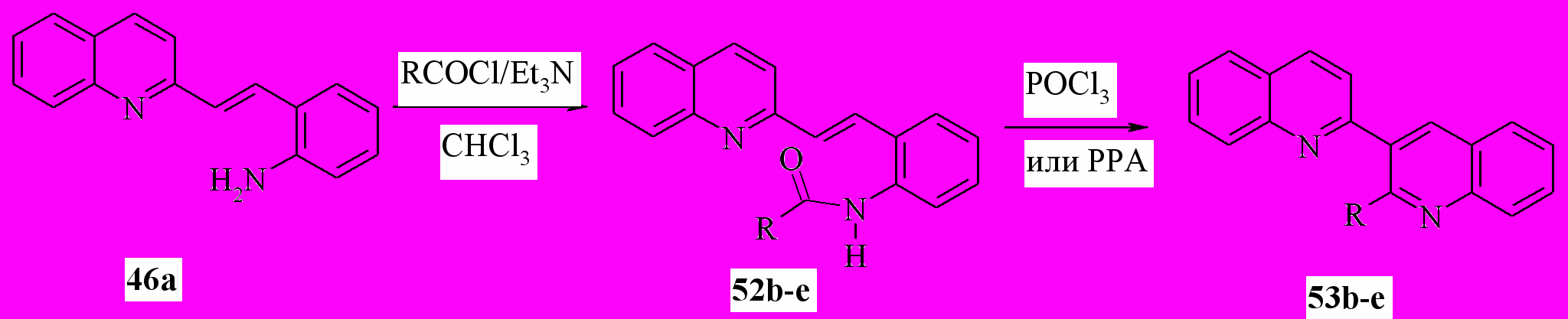
52,53b: R= Me, X=H; c: R=Pr, X=H; d: R=Bu, X=H; e: R= Ph, X=H
Таким образом, впервые реакция Робинсона была применена для синтеза бис и полигетероциклов.
Эту методологию удалось распространить на синтез тетрахинолинов. Для этого нами был получен диамид щавелевой кислоты 54. Его нагревание в ПФК приводит к тетрахинолину 55 с выходом 34%:
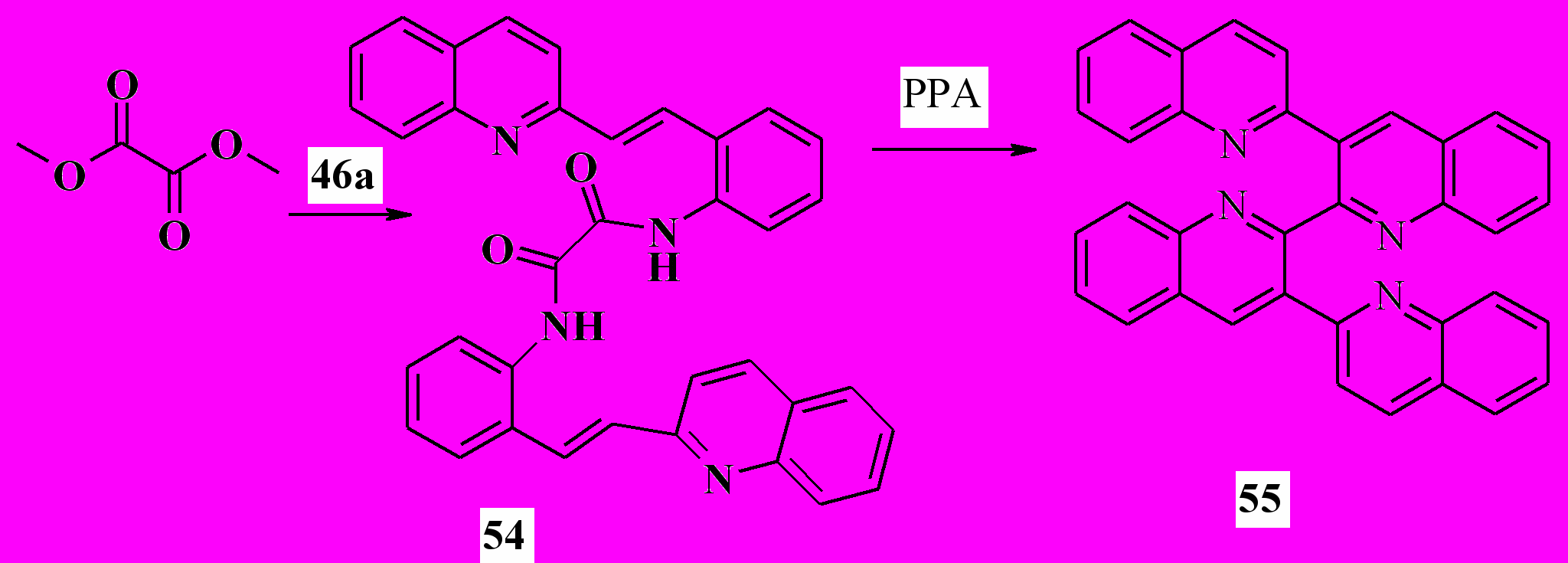
Далее, мы модифицировали этот метод, объединив реакции Робинсона и Вильсмайера. На первой стадии нарабатывается реагент Вильсмайера из ДМФА или диэтиламидов других карбоновых кислот, которым далее ацилируют амины 46-51:
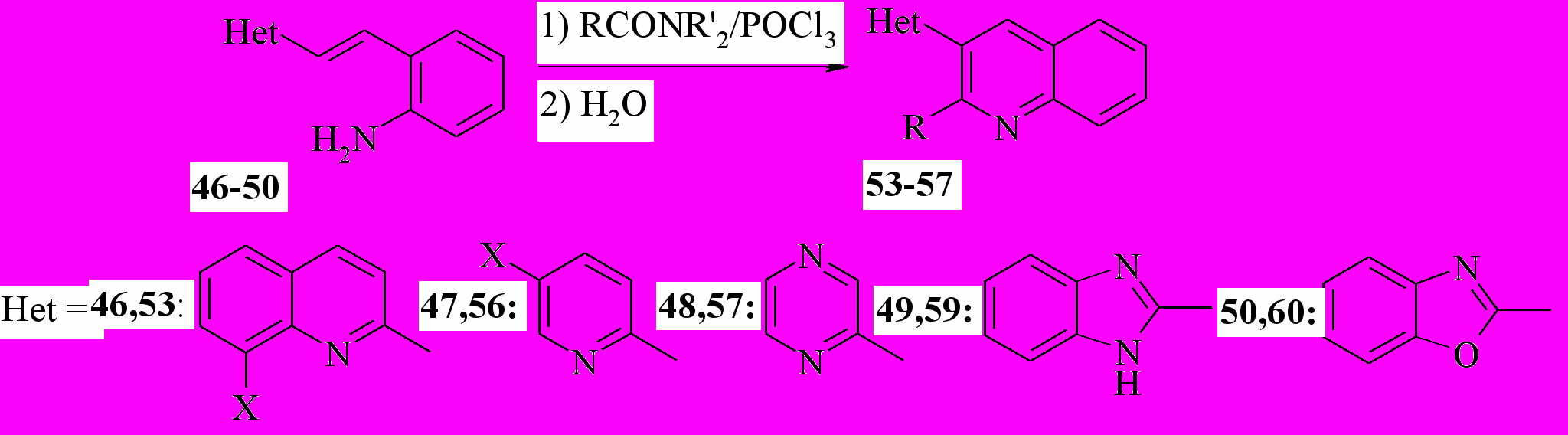
46a: X=H; b: X=NO2; с: X = Br; 53a: R = X = H; b: R = Me, X = H; c: R= Pr, X = H; d: R = Bu, X = H; e: R = Ph, X = H; f: R=H; X=NO2; g: X=Br; R = H; 47,56a: X=H; b: X=Br; c: X=Me;
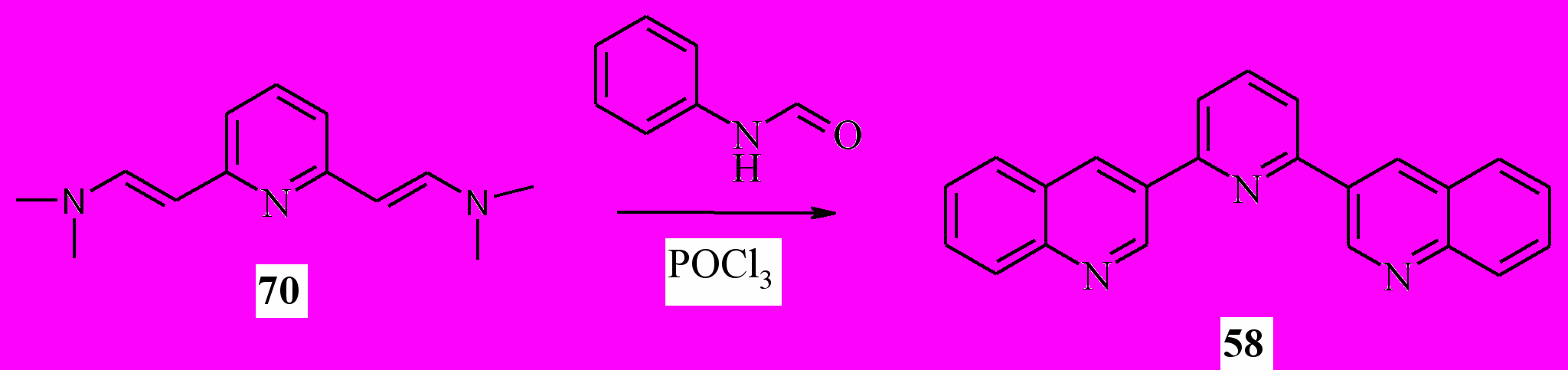
Далее, мы распространили эту методологию для синтеза других 3-гетарилхининолинов. Мы показали, что с помощью этой методологии можно синтезировать 3-(2-пиридил)хинолины (56a-c), 3-(2-пиразинил) хинолин (57), 3-(2-бензимидазолил)хинолин (59) и 3-(2-бензимидазолил) хинолин (60). Используя продукт конденсации по двум метильным группам колидина (51) с выходом 42% был получен трис гетероцикл 58:
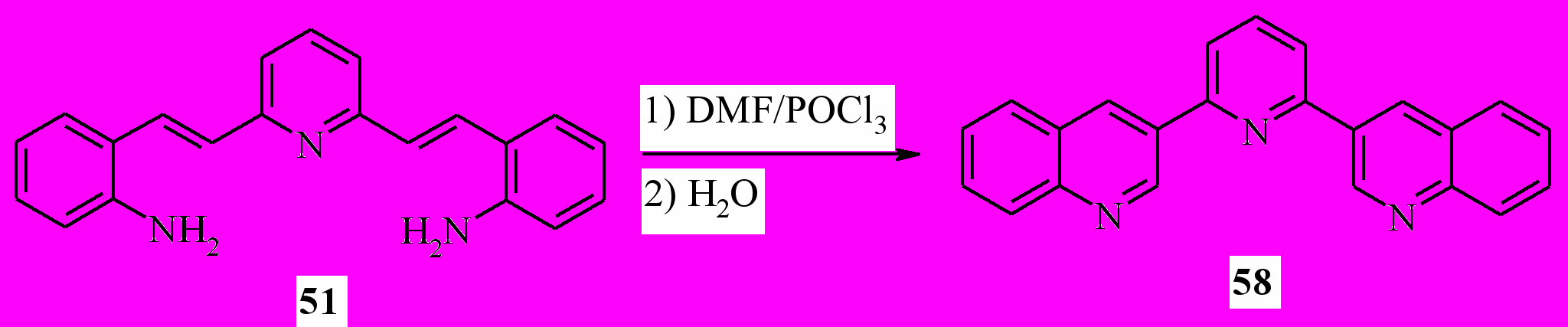
Таким образом, нам удалось разработать общий метод синтеза 3-гетарилхинолинов, основанный на комбинации реакций Робинсона и Вильсмайера.
Следующая часть нашей работы была посвящена исследованию возможности синтеза 1,2-дигидропроизводных 3-гетарилхинолинов из -гетарил-2-аминостиролов (46, 47). За основу была взята реакция конденсации -непредельных карбонильных соединений с альдегидами в присутствие DABCO (реакция Баилса-Хиллмана).
Мы предположили, что ее можно перенести на основания Шиффа, реализовать внутримолекулярный вариант и таким образом использовать для синтеза дигидропроизводных хинолинов. Реакцию проводили in one pot. Сначала в ТГФ смешивали карбонильное соединение с амином 46 или 47 и грели. При этом нарабатывалось основание Шиффа. Затем смесь охлаждали, добавляли DABCO и оставляли на 20-30 дней. В результате реакции с умеренным выходом 22-54% были получены 1’,2’-дигидропроизводные 2,3’-бихинолина (62) и 3-(2-пиридил) хинолина (63):
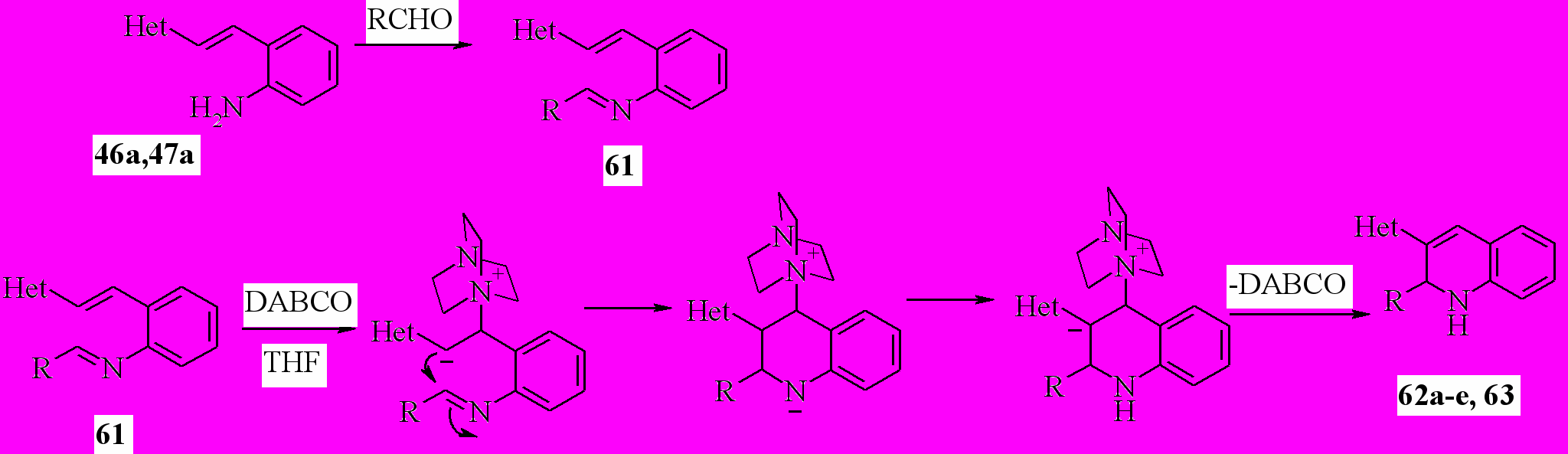
62: Het=2-хинолил; 62a: R= Ph; b: R = 1-C10H7; c: R = Me; d: R=Pr; e: R=Bu;
63: Het=2-пиридил, R=Ph
Таким образом, впервые была показана возможность применения реакции Баилса-Хиллмана для синтеза гетероциклических соединений.
До начала наших работ методов, включающих образование в 2,3'-бихинолинах связи С4’-С4’a, не существовало. В этой части работы мы разработали ряд таких методов. Сначала мы перенесли на производные 2,3’-бихинолина предложенный сравнительно недавно способ формирования замещенного хинолинового ядра, основанный на формилировании -хлоренаминов. Этим способом удалось получить 2’-хлор-2,3’-бихинолин с выходом 62%:
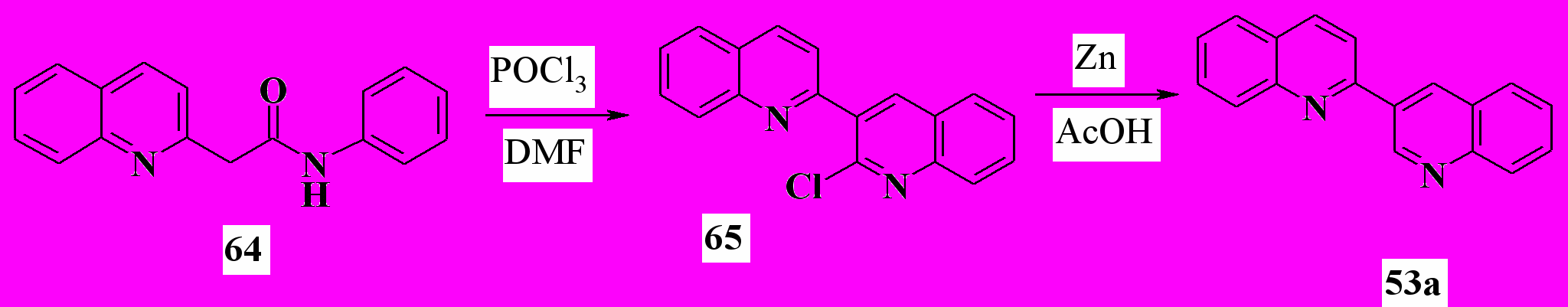
Восстановлением 65 цинком в уксусной кислоте с выходом 84% был получен 2,3’-бихинолин (53a).
За основу следующего метода мы взяли формилирование и ацилилирование енаминов соединениями, родственными реагентам Вильсмайера. Для чего по стандартной методике, исходя из хинальдина и диметилацеталя ДМФА, были синтезированы диметиламиновинилхинолины 66
Мы показали, что вместо диметилацеталя ДМФА, который является классическим реагентом для подобных синтезов, можно использовать его синтетический предшественник – аддукт ДМФА с диметилсульфатом в присутствие триэтиламина. Выход в этом случае ~70%, т.е. практически не отличается от предыдущего. При этом сокращается стадия синтеза. Полученные таким образом соединения 66 без очистки были пущены в реакцию с о-аминобензальдегидом и солью изатиновой кислоты. Выход 2,3’-бихинолинов составил 63-78%:
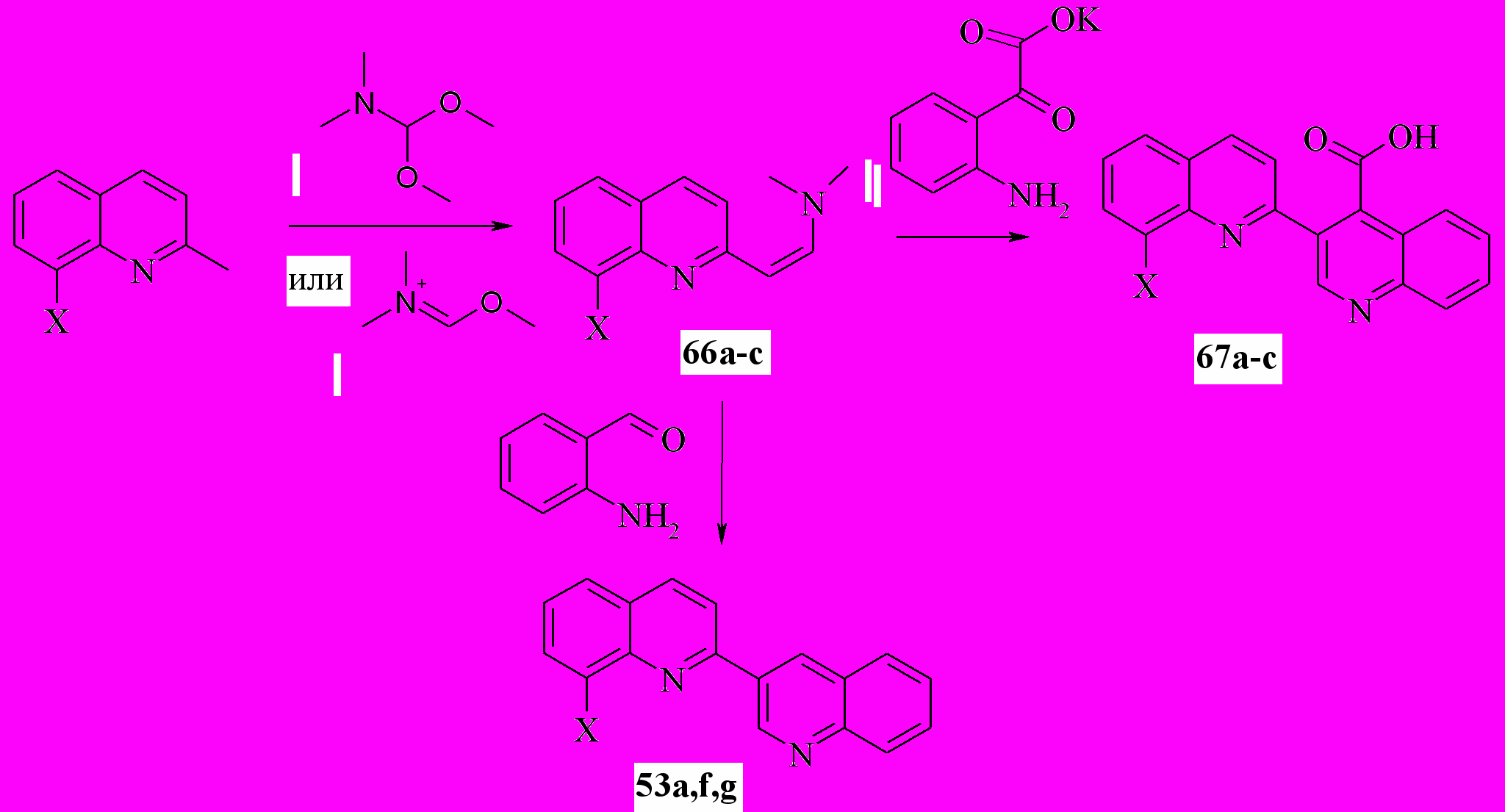
53a: X=H; f: X=NO2; g: X=Br; 66a: X=H; b: X=NO2; c:X=Br;
67a: X=H; b: X=NO2; с: X = Br;
Таким образом, нам удалось модифицировать известный метод, заменив альдегид хинолин 2-уксусной кислоты на более доступные диметиламиностиролы 66.
Далее мы показали, что 3-гетарилхинолины 53, 56,57,72 могут быть получены взаимодействием амидов кислот с соединениями 66, 68-71 в присутствие POCl3 Этот метод имеет преимущество по сравнению с описанными выше, так как не требует наличия в кольце дополнительной формильной группы и позволяет получать 2’-замещенные 2,3’-бихинолины. Выход составил 27-61%. Аналогично можно получить другие гетарилхинолины:

53a: R = X = H; b: R = Me, X = H; c: R= Pr, X = H; d: R = Bu, X = H; e: R = Ph, X = H; h: X = Br; R = H; 56a: X=Y=R=H; b: X=H,Y=Br,R=H; c: X=H,Y=Me, R=H; 57,72: X=R=H
Метод можно использовать и для синтеза трис гетероциклических систем:
Вероятно, реакция протекает по следующей схеме, включающей на первой стадии образование имидоил хлоридов, которые присоединяются по кратной связи енамина. Образовавшиеся соли иминов 73 циклизуется, давая дигидрохинолины 74, которые, теряя диметиламин, образуют 3-гетарилхинолины:
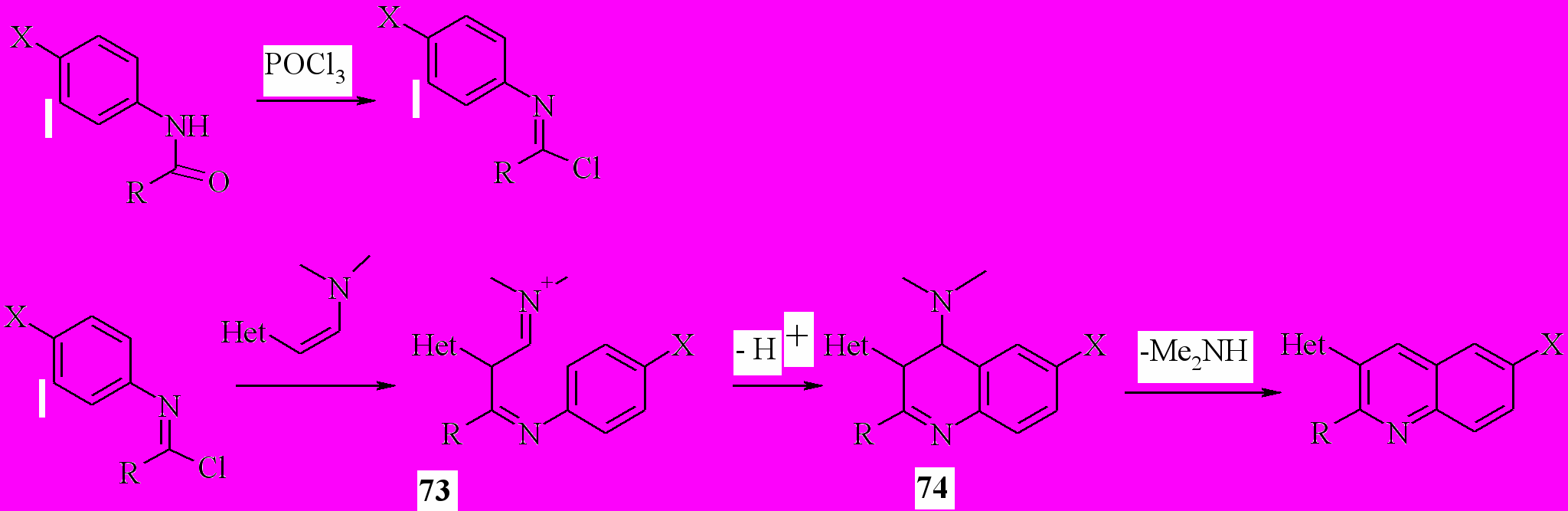
Наиболее низкий выход продукта наблюдается в случае форманилидов, что, вероятно, связано с низкой стабильностью соответствующих имидоил хлоридов.
3,3'-Бихинолин (72) был получен встречным синтезом из 3- бромхинолина, для чего пришлось модифицировать известную методику. Мы показали, что вместо палладиевой черни можно использовать 10%-ный Pd/C. Последний можно регенерировать, используя систему гидразин/КОН. При этом выход практически не изменился и составил 48%.
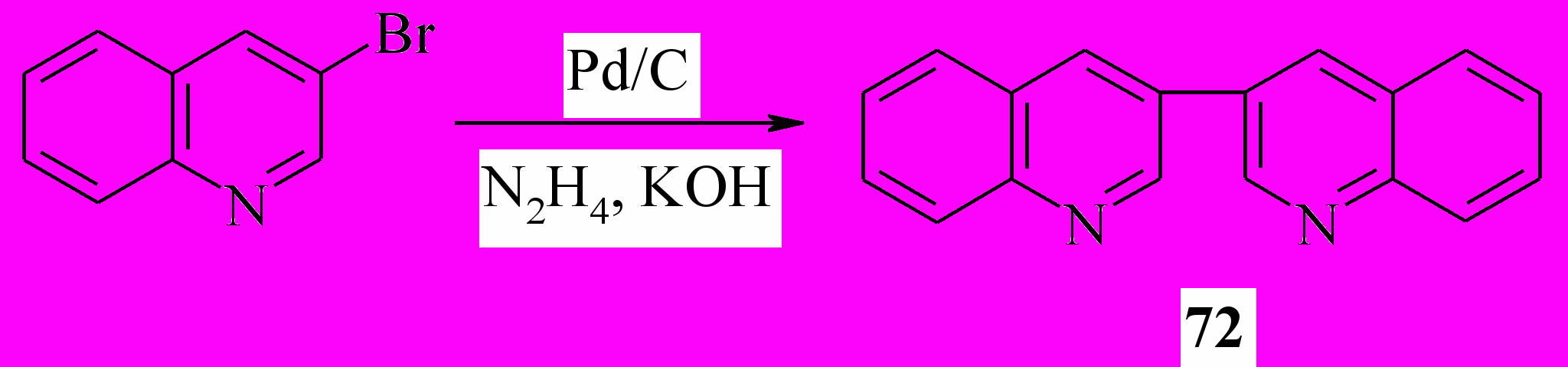
Далее, мы выяснили, что катализатор можно регенерировать в ходе реакции, постепенно добавляя к реакционной смеси раствор щелочи в гидразин-гидрате. Это позволило использовать каталитические количества палладия и многократно использовать катализатор. Однако выход в этом случае уменьшается до 32%.
В следующей части нашей работы мы реализовали еще один вариант процедуры Вильсмайера для синтеза 3-гетарилхинолинов. Для этого по стандартной методике были синтезированы соли 75, 76. Далее, реакцией соли 75 с хлориминиевыми солями в пиридине получали 2,3’-бихинолины (53) и 3-(2-пиридил)хинолин (56a):
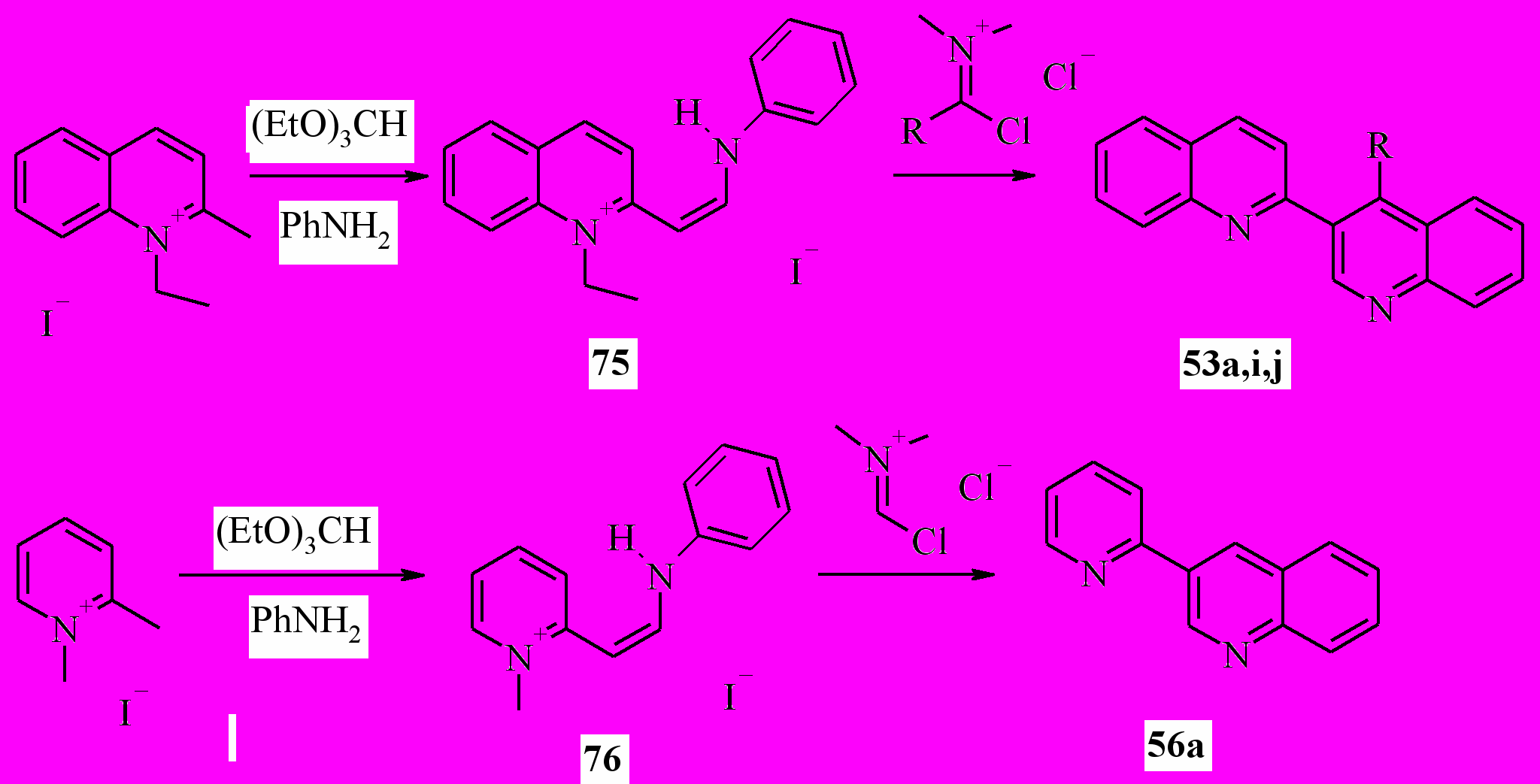
53a: R = H; i: R = Me; j: R = Ph;
Выход бихинолинов 53 составил 72-78%, пиридилхинолина 56a – 75%. Этот метод, в отличие от предыдущего, позволяет получить 4-замещенные 3-гетарилхинолины. Реакция, вероятно, протекает через следующую последовательность стадий (на примере 54a):
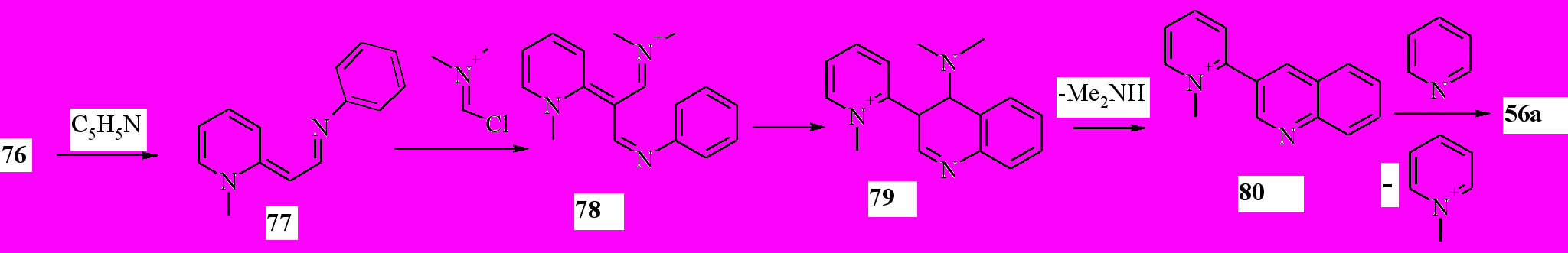
На первой стадии происходит депротонирование соли 76. Образовавшийся енамин 77 превращается в соль 78, которая циклизуется с образованием соли 79. Последняя теряет диметиламин, образуя соль 80, деметилирование которой пиридином приводит к 56a.
Далее мы распространили процедуру Вильсмайера на синтез 1,4-дигидропроизводных 3-гетарилхинолинов. В качестве модели нами были выбраны 1’,4’-дигидро-2,3’-бихинолины (82). Как следует из механизма на стр.27 реакция применима и для синтеза 82. В этой модификации в исходном соединении диметиламиногруппу соединения 66a заменили на H, Alk, Ar.
Нами установлено, что при кипячении 1 ммоль соединений 81, 1.1 ммоль форманилида или N-метилформанилида и 2 ммоль POCl3 в хлороформе в течение 2.5 ч и последующей обработкой водой и раствором аммиака образуются дигидропроизводные 82 с выходом 56-76%:
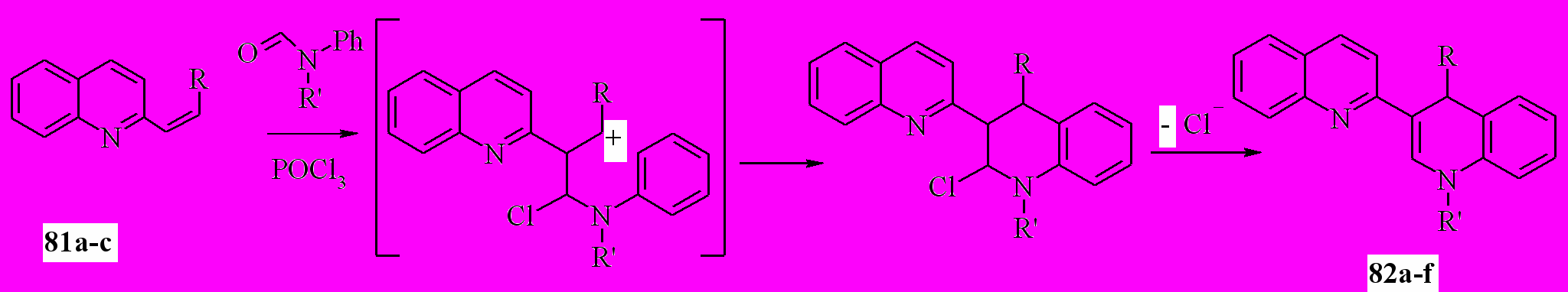
81a: R = H; b: R = Me; c: R = Ph; 82a: R = R’ = H; b: R = Me, R’ = H; c: R = Ph, R’ = H; d: R =H, R’ = Me; e: R = Me, R’ = Me; f: R = Ph, R’ = Me
Подобный подход был применен для синтеза полиядерных соединений – производных 1,3-диазапиренов ( 83). Выход составил 42-68%.

83a: R=H; b: R=Me; c: R=Ph
Завершая наши исследования применения процедуры Вильсмайера для синтеза 3-гетарилхинолинов, мы использовали ее для создания 2-замещенного хинолинового фрагмента. В качестве модели нами были выбраны 2,3’-бихинолины (53a,h). Задача представляет собой создание связей С2-С3 и С4-С4a. В качестве исходных были взяты анилиды хинолин-3-карбоновой и 6-бромхинолин-3-карбоновой кислот. Мы показали, что при последовательной реакции исходного анилида с SOCl2 в хлороформе, обработки реакционной смеси триэтиламином и далее кипячение с бутилвиниловым эфиром приводит к бихинолинам 53a,h с выходом 58 и 52% соответственно.
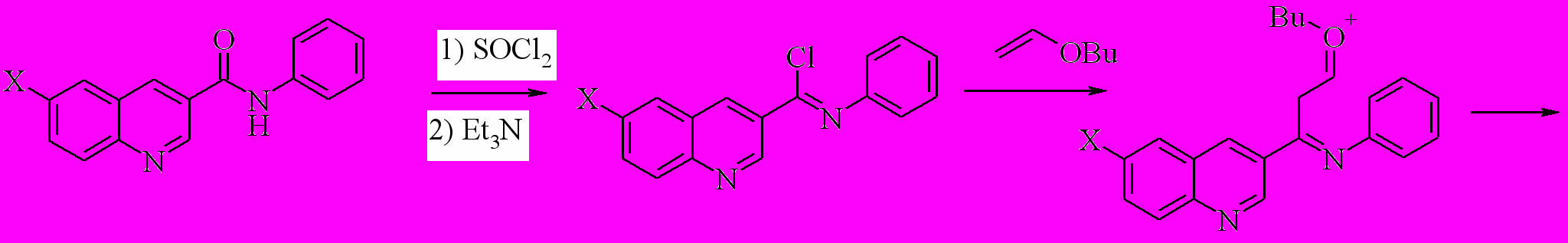
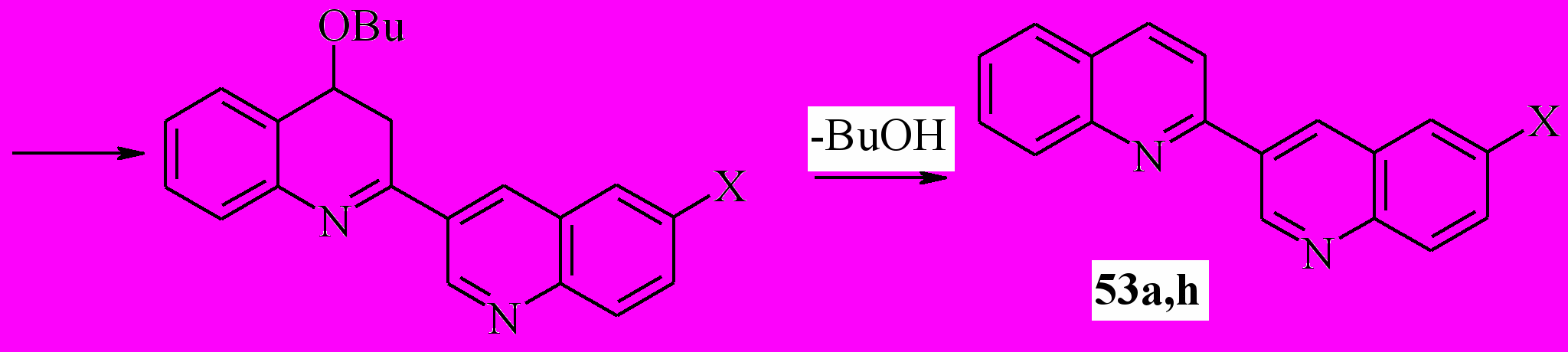
53a: R = Ph, X = H; h: X = Br; R = H;
Таким образом, на примере синтеза 3-гетарилхинолинов и их дигидропроизводных показаны синтетические возможности процедуры Вильсмайера для создания различных связей в различных хинолиновых ядерах бисгетероциклической системы (С1-С2 и С2-С3, С2-C3 и С4-С4а, С3-C4 и С4-С4а).
Ранее был разработан ряд методов синтеза 4'-хинолонов производных 2,3'-бихинолина – аналогов известных противомикробных препаратов. Одностадийных методов синтеза этих соединений, исходя из простых производных хинолина, известно не было. В этой части работы мы разработали такой метод синтеза, основанный на конденсации Клайзена.
Мы показали, что хинолоны 84 можно получить из хинальдина и метилового эфира N-алкил-N-формилантраниловой кислоты:
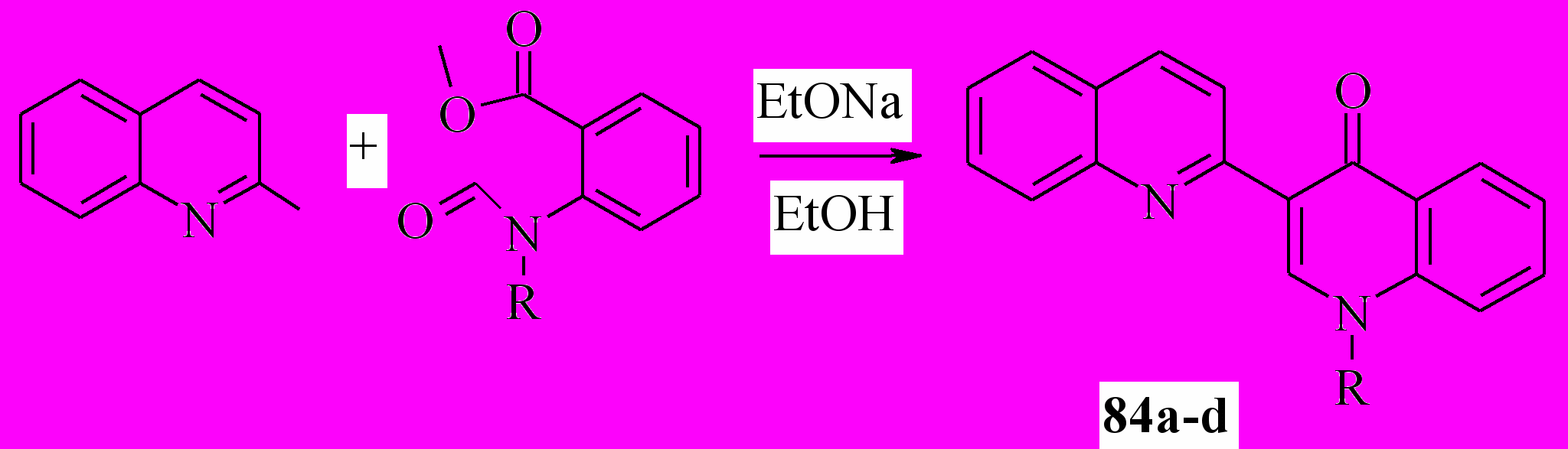
84a: R = Me; b: R = Et; c: R = Bu; d: CH2Ph
В случае алкильных заместителей на атоме азота выход практически не зависит от природы радикала и составляет 48-51%. В случае бензильного он значительно ниже 18%, что, вероятно, связано с побочным процессом дебензилирования.
Хинолоны 84 были получены и встречным синтезом через тионы 86, для чего был усовершенствован их синтез, используя следующую последовательность стадий: “защита” 1’-алкила в солях 85, путем их восстановления NaBH4 в дигидропроизводные 82 и далее, тиолирование последних элементной серой, как one pot-превращение:
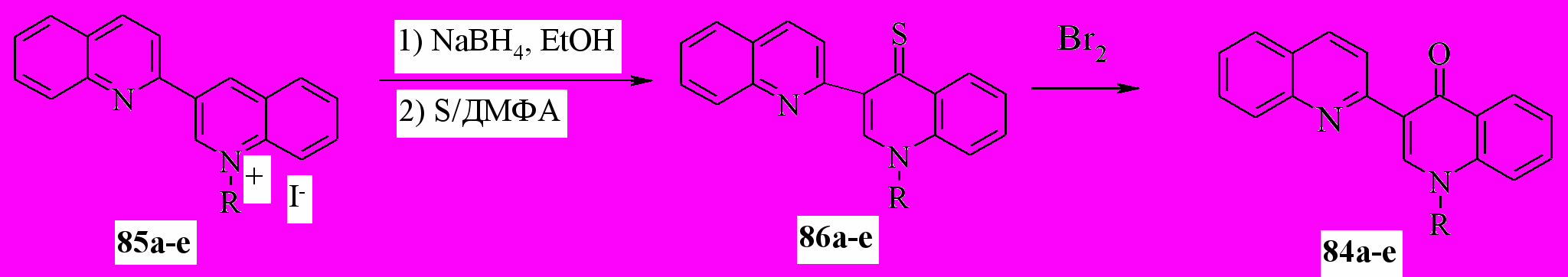
84,86а: R = Me; b: R = Et; c: R = n-Bu; d: R = CH2Ph; e: R = i-C5H11
Последующее окисление тионов 86, например, элементным бромом приводит к хинолонам 84а-e с выходом, близким количественному.
Ранее было изучено восстановление 2,3’-бихинолинов и показано, что оно протекает в две обратимые одноэлектронные стадии с образованием анион-радикала и дианона. Были получены некоторые данные по восстановлению кватернизованных 2,3’-бихинолинов (85). Региоселективность восстановления различными металлами определена не была. Поэтому данная часть работы была посвящена решению этой задачи.
Восстановление проводили, используя металлический цинк, литий и калий. Мы показали, что соли 85 с металлическим цинком в ТГФ образуют с практически количественным выходом смесь диастереомеров 88:
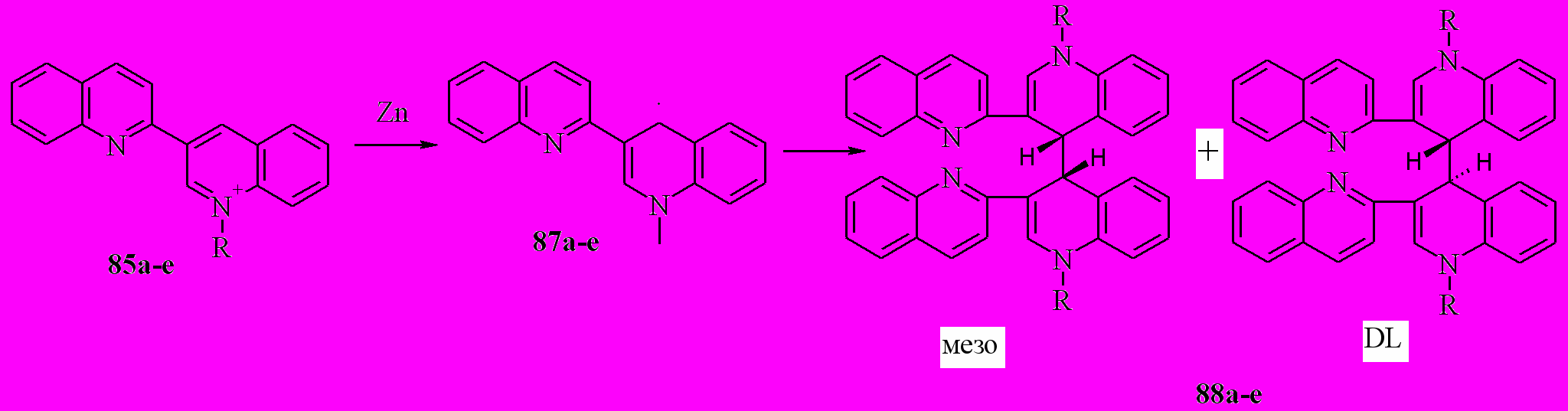
85,88a: R = Me; b: R = Et; c: R = Pr; d: R = CH2Ph; e: R = Bu;
Восстановление солей 85а-c,e металлическим калием в соотношении 1:3 в ТГФ приводит к образованию 1’-R-1’,4’-дигидро-2,3’-бихинолинов 82 с выходом 76-84 %:
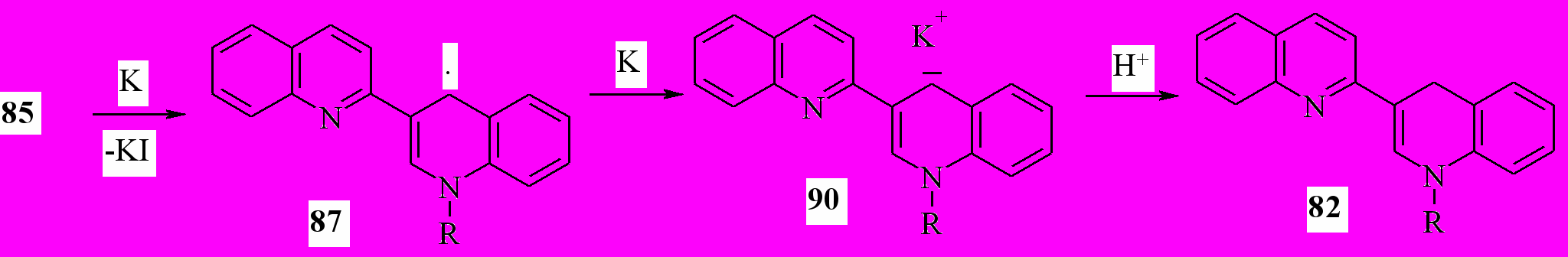
85a: R = Me; b: R = Et; c: R = Pr; e: R = Bu
Изменение региоселективности при переходе от цинка к калию можно объяснить следующим образом: калий, обладая более низким потенциалом ионизации, чем цинк способен восстановить радикал 87 до аниона 90, протонирование которого приводит к дигидропроизводному 82.
Восстановление соли 85d калием приводит к образованию после обработки водой с выходом 87 % 1’,4’-дигидро-2,3’-бихинолина (91). Этот результат можно объяснить, предположив механизм:
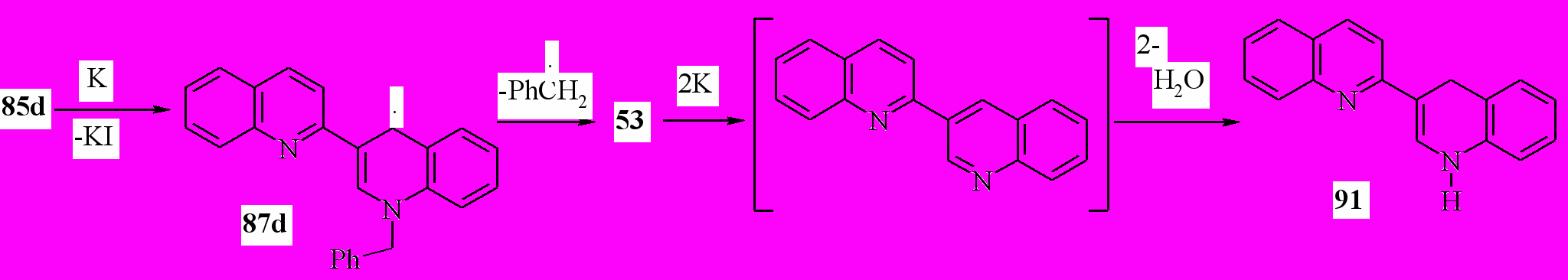
Казалось, что литий будет восстанавливать соль 85a аналогично калию. Неожиданно, в реакции соли 85а с 12-кратным избытком металлического лития в ТГФ была получена смесь 1’-метил-1’,4’-дигидро-2,3’-бихинолина (82d) и 1’,2’-дигидро-2,3’-бихинолина (90) с выходом 38 и 51 % соответственно:
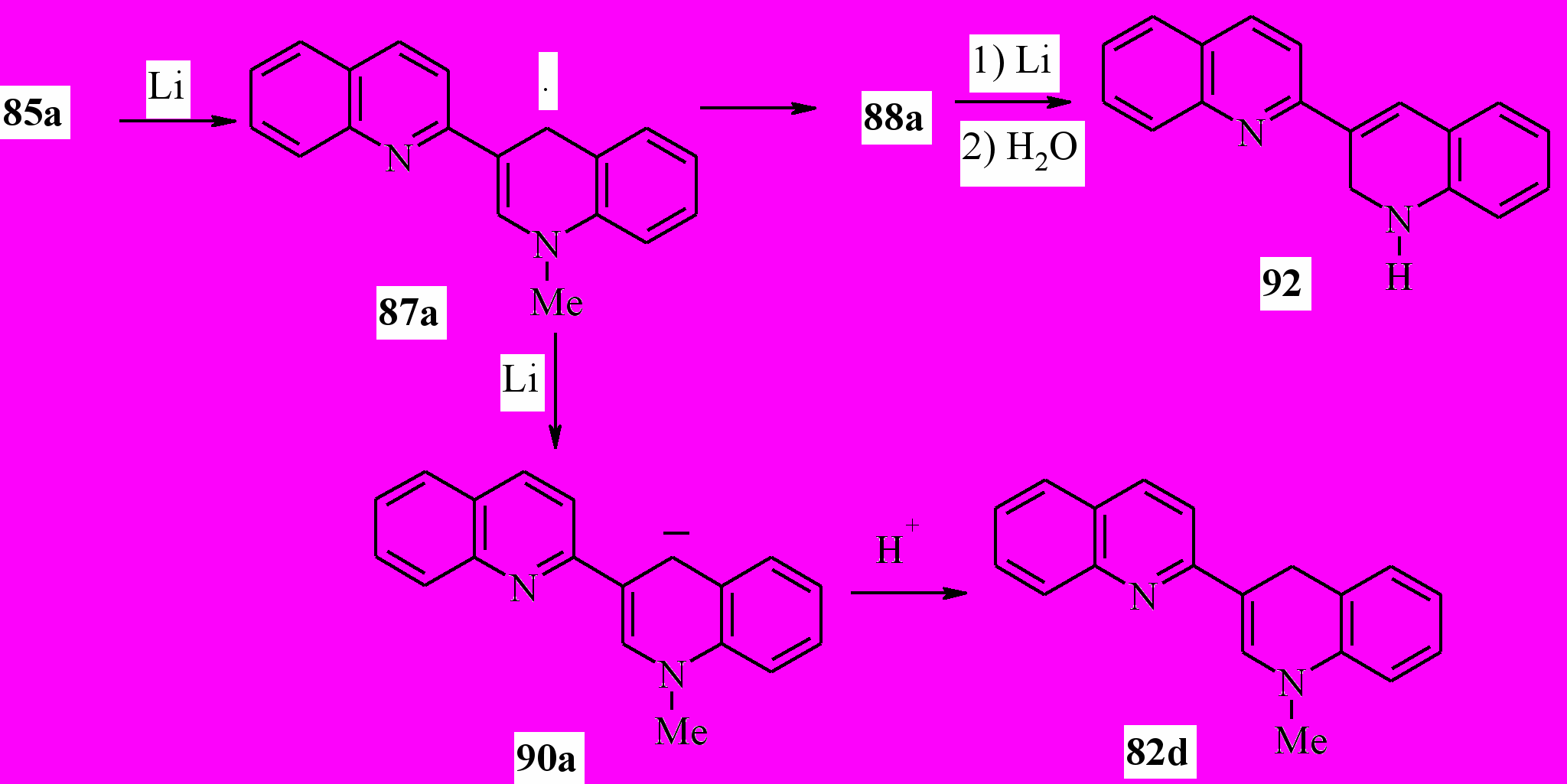
Вероятно, образующийся в ходе восстановления соли 85а радикал 87а частично восстанавливается до аниона 90, последующее протонирование, которого приводит к дигидропоизводному 82d. Частично происходит рекомбинация радикалов 87а с образованием димера 88a. Дальнейшее восстановление 88а с деметилированием приводит к соединению 92. Эта схема подтверждается тем, что восстановление соединения 88a при комнатной температуре 4-кратным избытком лития в абсолютном ТГФ в течение 1 ч приводит после обработки реакционной смеси водой к дигидропроизводному 92 с выходом 68 %
Таким образом, варьируя металл, удалось изменить региоселективность восстановления солей 85 и разработать методы синтеза различных частично гидрированных производных 2,3'-бихинолина.
Следующая часть работы была посвящена реакции дигидропроизводных 2,3'-бихинолина с LDA. Неожиданно, основным продуктом реакции 82d с LDA в ТГФ оказался 1,1’-диметил-3,3’-ди(3-хинолил)-1,1’,2,2’-тетрагидро-2,2’-бихинолин (89а). Выход 82%. В качестве побочных продуктов образуются 88а (выход 7%) и 1,1’-диметил-3,3’-ди(3-хинолил)-1,1’,2,4’-тетрагидро-2,4’-бихинолина (89b) (выход 5%):
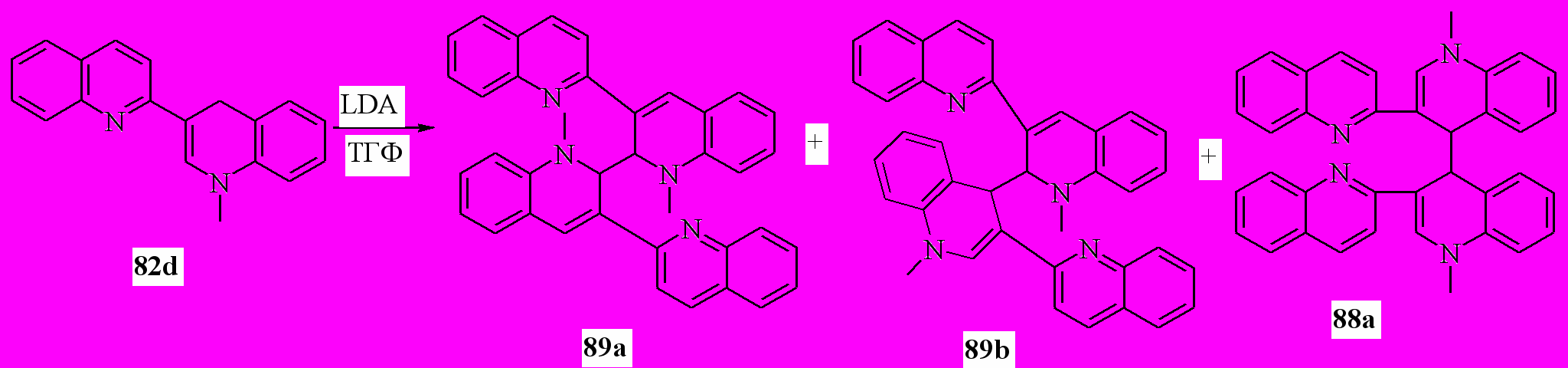
Вероятно, механизм реакции аналогичен, описанному ранее для взаимодействия дианиона 2,3’-бихинолина с литийорганическими соединениями в ТГФ. В отсутствие внешнего нуклеофильного реагента в качестве такового выступает одна из молекул аниона 90
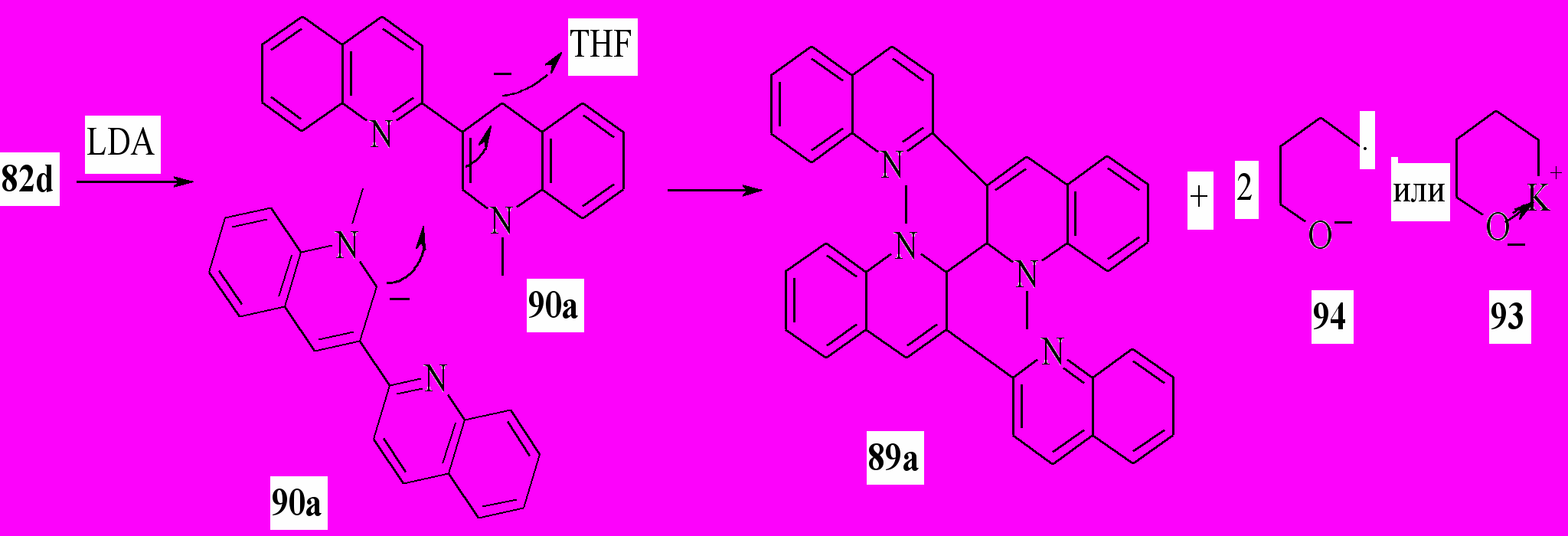

Мы установили, что если реакционную смесь после обработки водой покипятить в течение 0.5 ч, то единственным продуктом реакции будет соединение 88а. Этот результат можно объяснить тем, что димеры 89a и 89b при нагревании образуют свободный радикал 87, рекомбинация которых по положению с максимальной спиновой плотностью приводит к соединению 88. Аналогичная перегруппировка наблюдается при кипячении соединения 89a в бензоле.
Таким образом, разработан метод синтеза ранее неизвестных гетероциклических аналогов тетракарбонильных соединений - 1,1’-диалкил-3,3’-ди(3-хинолил)-1,1’,2,2’- тетрагидро-2,2’- бихинолинов и открыта их перегруппировка в 1,1’-диалкил-3,3’-ди(3-хинолил)-1,1’,4,4’- тетрагидро-4,4’- бихинолины.
В следующей части работы изучились реакции производных тетрахинолина 88 с нуклеофильными и электрофильными реагентами.
Мы показали, что реакция соединений 88 с литийорганическими соединениями в ТГФ при комнатной температуре, приводит к образованию дигидропроизводных 95 с количественным выходом. Вероятно, реакция протекает аналогично образованию 89a:
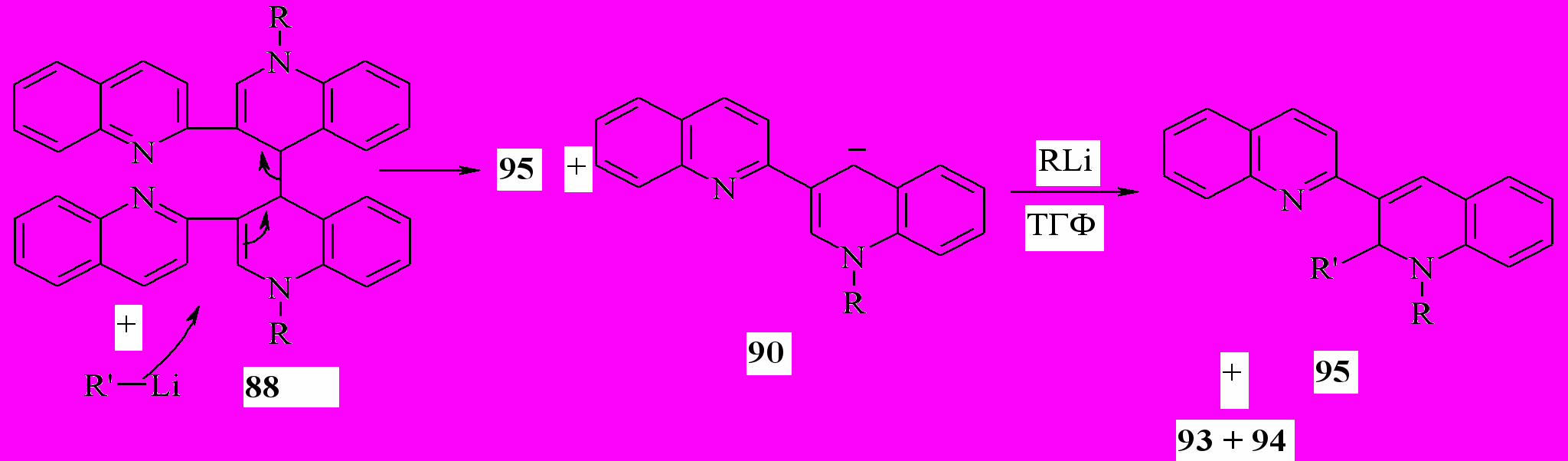
95a: R=R’=Me; b: R=Me, R’=Ph; c: R=Et, R’=Me; d: R=Et, R’=Ph
На первой стадии литийорганическое соединение выступает в качестве нуклеофильного реагента по отношению соединению 88. В качестве уходящей группы выступает анион 90. В результате нуклеофильного замещения с аллильной перегруппировкой образуется 95 и анион 90. Реакция последнего с литийорганическим соединением, как было показано ранее для реакции 1’-R-1’,4’-дигидро-2,3’-бихинолинов с литийорганическими соединениями. В результате этой реакции, как и на первой стадии, образуются соединения 95.
Аналогичным образом протекает реакция соединений 88a с реактивами Гриньяра. Выход соединений 95 в этом случае также количественный.
Если направление реакций 88a с нуклеофильными реагентами, во всяком случае, на первой стадии (присоединение по Михаэлю), предсказуемы, то с электрофильними реагентами их направление не очевидно. Поэтому следующая часть работы была посвящена их исследованию.
Мы предположили, что вследствие наличия в молекулах исследуемых аналогов тетракарбонильных соединений 88 хорошего электрофуга в положении 4, они могут реагировать с электрофильными реагентами не только по атомам азота и атому углерода в положении 3, но и в 2 с образованием в результате электрофильного замещения с аллильной перегруппировкой дигидропроизводных (96) и солей бихинолиния 85:
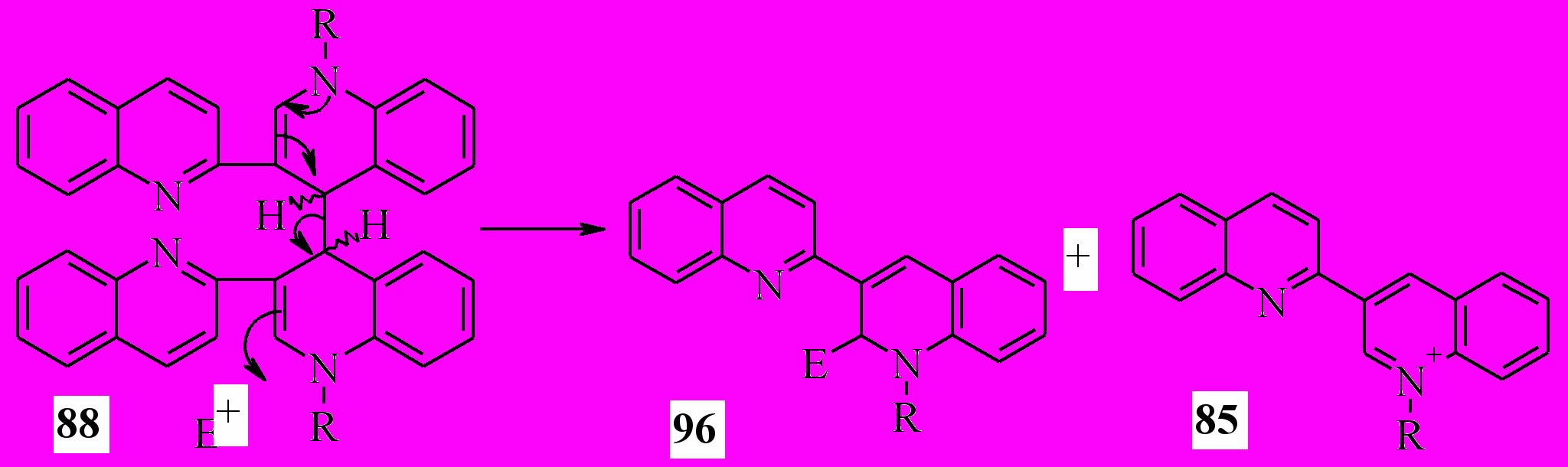
Реакции 88a-e с хлором бромом и йодом проводили при комнатной температуре в четыреххлористом углероде. Единственными продуктами реакции оказались соли 85. Вероятно, реакция протекает по схеме:
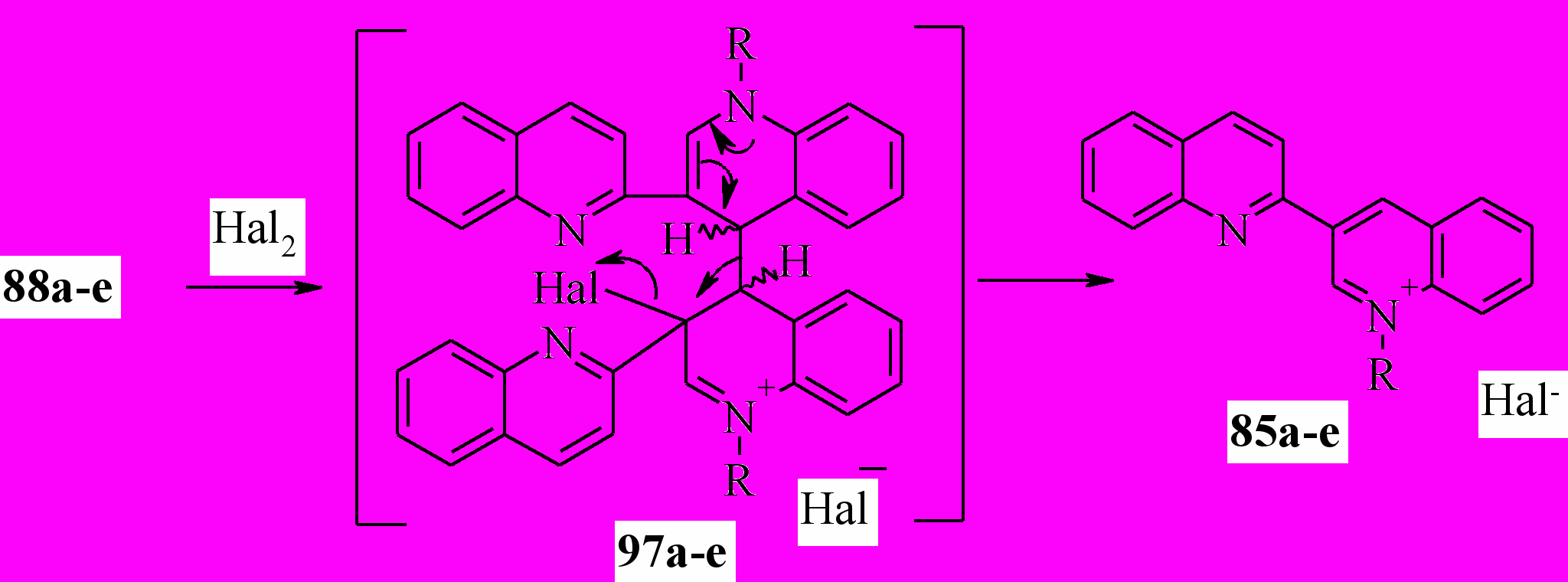
85, 97a: R = Me; b: R = Et; c: R = Pr; d: R = CH2Ph; e: R = Bu; Hal = Cl, Br;
Кипячение терахинолинов 88a-e со спиртовыми растворами HCl, HBr и n-толуолсульфокислоты приводит к образованию 82a,g-j и солей 85a-e в равном соотношении. Суммарный выход соединений 85 и 82 близок количественному. Реакция, вероятно, включает следующую последовательность стадий. Сначала, в результате электрофильного замещения с аллильной перегруппировкой образуются 1’,2’-дигидро-2,3’-бихинолины, которые, как известно, легко перегруппировываются в 1’,4’-дигидропроизводные 82:
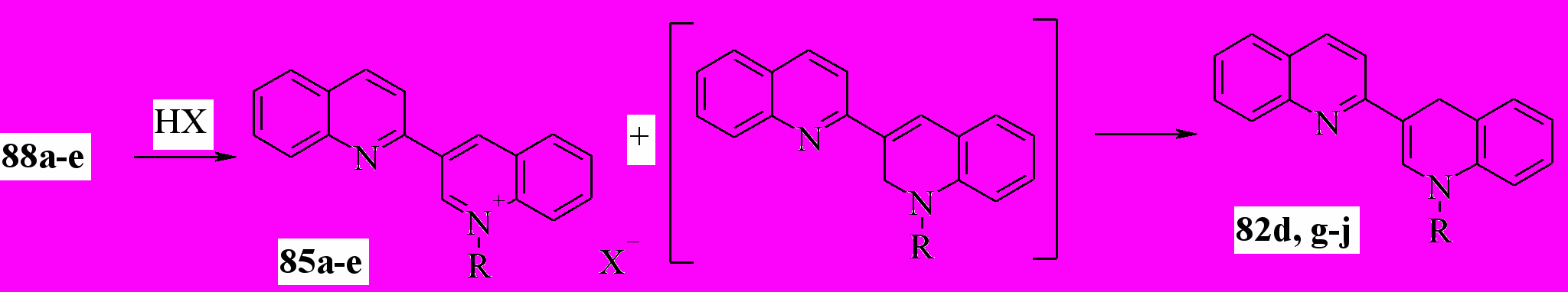
82d: R = Me; g: R = Et; h: R = Pr; i: R = Bu; j: R = CH2Ph; X = Cl, Br, OTs
Можно было ожидать, что реакция соединений 88 с хлорангидридами кислот будет протекать аналогично. В этом случае в качестве продуктов реакции должны образовываться соли 85 и дигидропроизводные 98. Действительно, при взаимодействии соединений 88а, b, e с этилхлорформиатом одним из продуктов реакции являются соли 85a, b, e (выход близок к количественному). Неожиданно, в качестве второго продукта был выделен эфир 99a с выходом 44 % (не зависимо от радикала при атоме азота в положении 1’). Вероятно, реакция включает промежуточное образование дигидропроизводных 98, которые, в результате окислительного дезалкилирования образуют бихинолины 99:
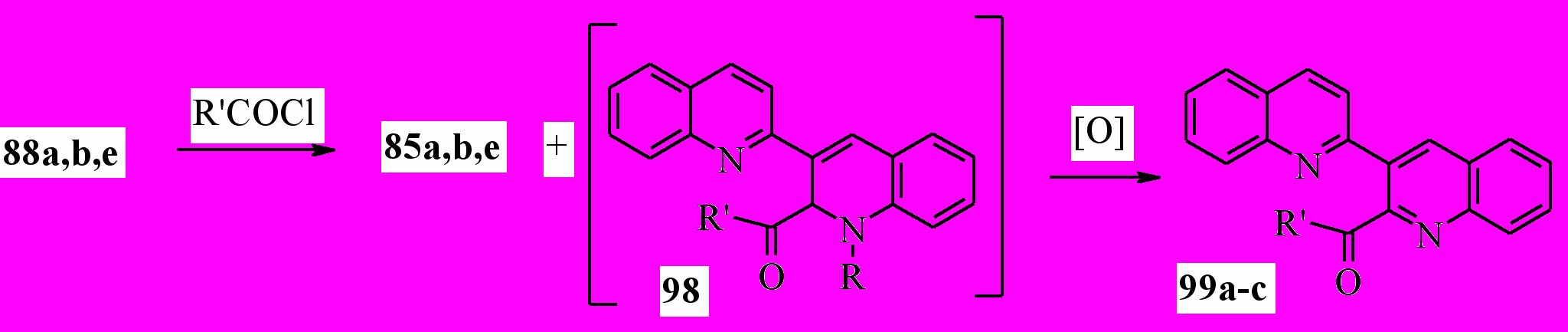
85, 88a: R = Me; b: R = Et; e: R = CH2Ph; 99a: R = OEt; b: R = Me; c: R = Ph
Реакция аналогично протекает с хлорангидридами других кислот.
Таким образом, в этой части работы исследованы реакции тетрахинолинов 88 с галогенами и минеральными кислотами. Открыта реакция ацилирования дигидропроизводных 2,3’-бихинолина в положение 2’, что позволило разработать методы синтеза кетонов, производных 2,3’-бихинолина и эфира 2,3’-бихинолин-2’-карбоновой кислоты. Показано, что с металлоорганическими соединениями образуются 2'-R-1',2'-дигидро-2,3’-бихинолины.
В следующей части нашей работы мы изучили возможности циклизации дигидробихинолинов и солей бихинолиния. Известно, что сера реагирует с енаминами с образованием соответствующих тиокарбонильных соединений. Мы предположили, что взаимодействие с серой диметилпроизводного 95a приведет через соответствующий енамин и тиоформильное производное к цвиттер-иону 100. Действительно, в реакции соединения 95a с серой образуется продукт 100 с выходом 73%:
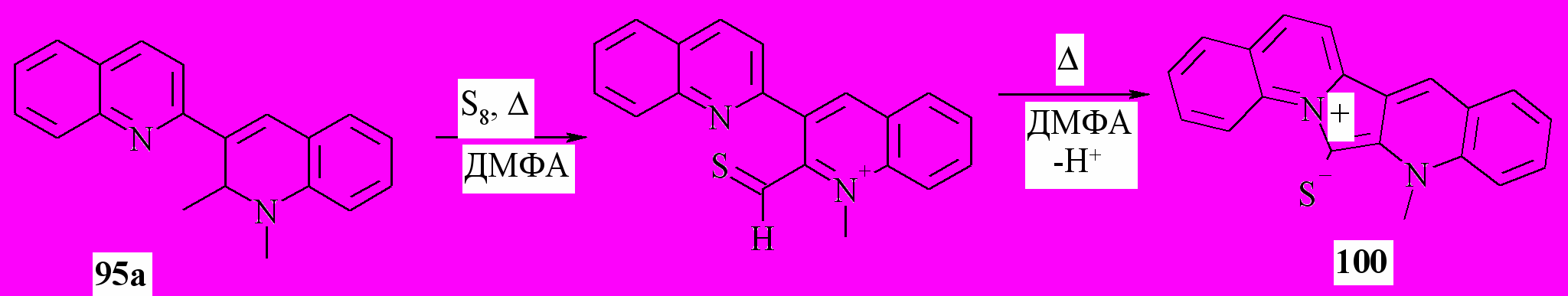
Далее, в реакции дигидробихинолина 82f с серой также можно было ожидать циклизации. Образовавшийся азиниевый катион в данных условиях может нуклеофильно тиолироваться в тиоамид 101:
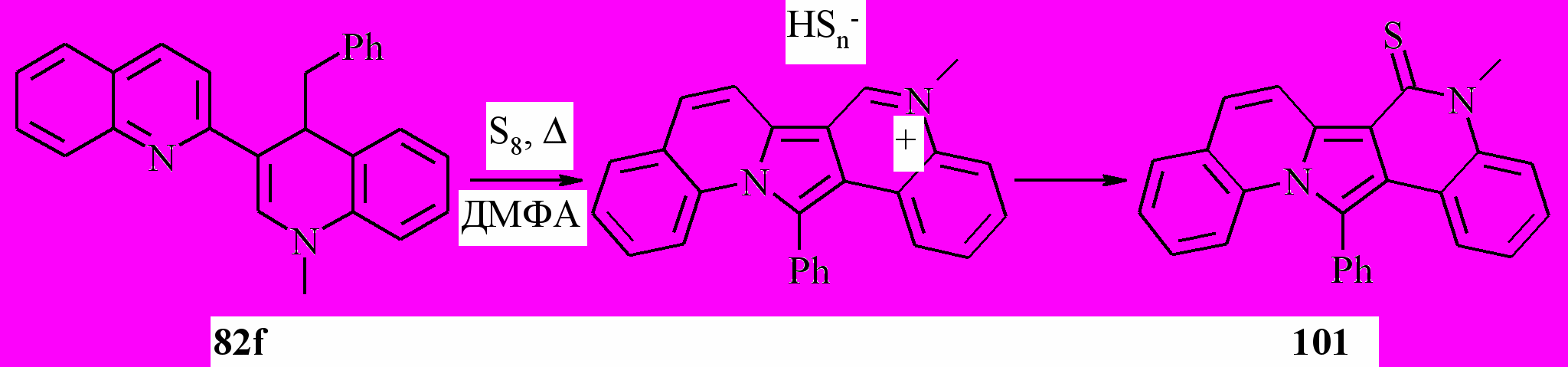
Мы показали, что соединение 101 образуется в данной реакции с выходом 62%.
Таким образом, мы продемонстрировали возможность замыкания дополнительного пятичленного цикла, используя атом азота в положении 1, создавая тиокарбонильную группу в положениях 2' или 4'.
4. Биологическая активность замещённых (O,N)гетеро-1,3-диенов, их нециклических и кольчатых аза-аналогов
В результате биологических испытаний полученных нами соединений была обнаружена выраженная противомикробная активность, а также анальгетическое действие при низкой токсичности. Острая токсичность испытанных соединений находится, в основном, в пределах от 400 до 1000 мг/кг и более при внутрибрюшинном пути введения. Таким образом, большинство изученных соединений являются мало токсичными или практически не токсичными и имеют в этом преимущество перед препаратами сравнения биологической активности. Противомикробную активность синтезированных соединений по отношению к эталонным штаммам кишечной палочки Escherichia coli M17 и золотистого стафилококка Staphylococcus aureus P-209 определяли стандартным методом двукратных серийных разведений в мясо-пептонном бульоне при бактериальной нагрузке от 250 тысяч до 5 млн микробных единиц в 1 мл раствора. Бактериостатический эффект наиболее активных соединений сравнивали с действием применяемых противомикробных препаратов – этакридина лактата и современных антибактериальных препаратов группы 4-оксохинолин-3-карбоновой кислоты – флумеквина, оксолиниевой, налидиксовой кислот и норфлоксацина. Наибольшим противомикробным (бактериостатическим) эффектом обладают галогенпроизводные 1,3,4,6-тетракарбонильных соединений, амиды ацилпировиноградных кислот и замещённые индолиноны, активность которых (диапазон МИК от 0.125 до 1000 мкг/мл) находится на уровне применяемых в медицине препаратов (МПК 0.06 – 256 мкг/мл). Анальгетическую активность исследовали по методу "горячей пластинки" на белых мышах массой 16-24 г при внутрибрюшинном введении соединений в дозах 50 мг/кг в виде взвеси в 2% крахмальной слизи. Заметное противовоспалительное и анальгетическое действие характерно для разнообразных ацилпирувамидов. Все исследованные соединения являются малотоксичными или практически не токсичными и действуют в гораздо меньших эквитоксических дозах, чем препараты сравнения, что может быть использовано в целенаправленном поиске эффективных лекарственных средств среди (O,N)гетеро-1,3-диенов, их аналогов и производных.
***
Таким образом, в ходе проделанной работы нам удалось разработать и усовершенствовать значительное количество методов синтеза соединений, содержащих (O,N)гетеро-1,3-диеновое звено, показать общность методов применяемых для синтеза соединений алифатического и гетероциклического ряда. Расширены области применения некоторых классических синтетических методов, таких как конденсация Клайзена, реакция Вильсмайера, Баилса-Хиллмана и др. Были обсуждены особенности строения и реакции большого ряда (O,N)гетеро-1,3-диенов, их взаимопревращения и биологическая активность.
Выводы
- Разработано новое перспективное научное направление в области химии (O,N)-гетеро-1,3-диенов, в основе которого лежит создание новых методов синтеза гетеродиеновых структур с открытой цепью, их кольчатых аналогов, в том числе аннелированных и бис-гетероциклических систем. Обосновано представление об общности синтеза гетеро-1,3-диенов для получения разнообразных гетероциклов и модификации их структуры введением активированного окса(аза)-1,3-диенового звена.
- Показано, что активированные (O,N)-гетеро-1,3-диены и, в первую очередь, окса-1,3-диены представляют собой активные структурные блоки или синтетические эквиваленты синтонов, с помощью которых имеется возможность построения разнообразных окса- и азагетероциклических систем и открываются перспективы модификации молекул гетероциклов.
- Разработан ряд принципиально новых, препаративно удобных методов получения разнообразных ациклических и кольчатых систем, содержащих фрагменты активированных (O,N)гетеро-1,3-диенов, в том числе аннелированных и бис-гетероциклических производных с различными заместителями в линейном и циклическом звеньях молекул.
- Изучено строение линейных и циклических производных (O,N)гетеро-1,3-диенов и выявлена склонность к кето-енольным, амино-иминным цепным и кольчато-цепным таутомерным превращениям, способность образовывать моно-, бис-хелатных ансамблей, что позволило прогнозировать направление химических превращений.
- Определена региоселективность реакций активированных акцепторами (O,N)гетеро-1,3-диенов (например, ацилметиленоксиндолов) с N- нуклеофилами. Показано, что при переходе от кетонов к сложным эфирам изменяется направление атаки нуклеофила с β- на α-атом углерода ненасыщенного звена.
- На примере синтеза 3-гетарилхинолинов и их дигидропроизводных показаны синтетические возможности процедуры Вильсмайера для создания различных связей в различных хинолиновых ядрах бисгетероциклической системы (С1-С2 и С2-С3, С2-C3 и С4-С4а, С3-C4 и С4-С4а). На основе чего, разработано 6 новых методов синтеза 3-гетарилхинолинов и их дигидропроизводных.
- Впервые реакция Баилса-Хиллмана применена для синтеза гетероциклических соединений, на основании чего, разработан новый метод синтеза 1,2-дигидропроизводных 3-гетарилхинолинов.
- На основе ранее неизвестных реакций ацилирования хлорангидридами кислот и алкилирования(арилирования) металлоорганическими соединениями 1,1’-диалкил-3,3’-ди(3-хинолил)-1,1’,2,2’-тетрагидро-4,4’- бихинолинов разработаны методы синтеза 2'-замещенных 2,3'-бихинолинов.
- Изучена биологическая активность соединений, содержащих (O,N)гетеро-1,3-диеновое звено, их циклических аналогов и продуктов превращений, а также близких соединений, имеющих характеристические фармакофорные группы, определяющие биологический эффект. В результате фармакологических испытаний синтезированных веществ, в том числе некоторых гидразидов, выявлено более 60 веществ, обладающих противомикробной, противовоспалительной и анальгезирующей активностью. Обнаружены соединения с противомикробным действием, которые по своему эффекту превосходят препараты, применяющиеся в настоящее время в клинической практике, что предполагает актуальным их дальнейшие фармакологические исследования.
Основное содержание работы изложено в следующих публикациях: