
«Применение ит в молекулярной генетике»
| Вид материала | Реферат |
- Научный семинар по молекулярной экологии и молекулярной биологии рспространенных заболеваний, 79.62kb.
- «Применение Интернет-ресурсов в молекулярной генетике», 114.91kb.
- Рабочая программа дисциплины «молекулярная генетика» Код дисциплины по учебному плану, 112.43kb.
- Учебно-методический комплекс дисциплина: молекулярная биология и медицинская генетика, 2387.39kb.
- Научный совет по генетике и селекции ран, 59.12kb.
- Применение модульной технологии при обучении генетике в старших классах, 188.31kb.
- Рабочая программа по медицинской генетике Для специальности n- 040200 «Педиатрия» Квалификация, 180.42kb.
- Исследование выполняется на базе кафедры биохимии и молекулярной биологии и нил молекулярной, 19.33kb.
- Молекулярно-генетическая характеристика индивидуального района интеркалярного гетерохроматина, 399.53kb.
- Курс молекулярной физики, 48.83kb.
БЕЛОРУССКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
Выпускная работа по
«Основам информационных технологий»
Магистрант кафедры генетики
Чернова Александра Игоревна
Руководители:
профессор Титок Марина Алексеевна,
ассистент Шешко Сергей Михайлович
Минск – 2008 г.
Оглавление
- Список обозначений ко всей выпускной работе…………………………….3
- Реферат на тему «Применение ИТ в молекулярной генетике»………...…..4
- Введение………………………………………………………………...…4
- Глава 1 (способы применения IT в молекулярной генетике.)……...…..8
- Применение IT для поиска необходимой информации………..…8
- Применение IT для проведения молекулярно-генетических исследований…………………………………………………………14
- Применение IT для поиска необходимой информации………..…8
- Глава 2 (Применение IT в исследованиях по подготовке магистерской диссертации)………………………………………………………..…….19
- Глава 3 (Заключение)……………………………………………….…..20
- Введение………………………………………………………………...…4
- Список литературы к реферату……………………………………………...21
- Интернет ресурсы в предметной области исследования..……………..…..23
- Действующий личный сайт в WWW (гиперссылка)………………..……..24
- Граф научных интересов………………………………………………..…...25
- Презентация магистерской (кандидатской) диссертации…………..…..…26
- Список литературы к выпускной работе…………………………………...27
1Список обозначений ко всей выпускной работе
IT, ИТ – информационные технологии
2Реферат на тему «Применение ИТ в молекулярной генетике»
2.1 Введение
Молекулярная генетика, раздел генетики и молекулярной биологии ставящий целью познание материальных основ наследственности и изменчивости живых существ путём исследования протекающих на субклеточном, молекулярном уровне процессов передачи, реализации и изменения генетической информации, а также способа её хранения.
Молекулярная генетика выделилась в самостоятельное направление в 40-х гг. 20 в. в связи с внедрением в биологию новых физических и химических методов (рентгеноструктурный анализ, хроматография, электрофорез, высокоскоростное центрифугирование, электронная микроскопия, использование радиоактивных изотопов и т. д.), что позволило гораздо глубже и точнее, чем раньше, изучать строение и функции отдельных компонентов клетки и всю клетку как единую систему. За свою недолгую историю молекулярная генетика достигла значительных успехов, углубив и расширив представления о природе наследственности и изменчивости, и превратилась в ведущее и наиболее быстро развивающееся направление генетики. Молекулярная генетика изучает молекулярные основы генетических процессов как у низших, так и у высших организмов и не включает частной генетики прокариот, занимающей видное место в генетике микроорганизмов.
Любую из современных наук сегодня нельзя представить без применения информационных технологий. Молекулярная генетика не стала исключением. С новыми методами в биологию пришли новые идеи физики и химии, математики и кибернетики. Современная биология стала производителем беспрецедентно огромных объемов экспериментальных данных, осмысливание которых невозможно без привлечения современных информационных технологий и эффективных математических методов анализа данных и моделирования биологических систем и процессов.
Прогресс человечества в 21 веке будет неразрывно связан с развитием и взаимодействием молекулярной биологии, генетики и информатики. Ответы на многие глобальные вызовы, стоящие перед современной цивилизацией, критическим образом зависят от развития этих наук, их взаимодействия и использования их достижений.
В ответ на эту острую потребность возникает новая наука - информационная биология. Объектами исследований информационной биологии являются генетические макромолекулы - ДНК, РНК, белки, фундаментальные генетические процессы - репликация, транскрипция, трансляция, генетические сети, функционирование которых обеспечивает выполнение всех функций организмов.
Генные сети
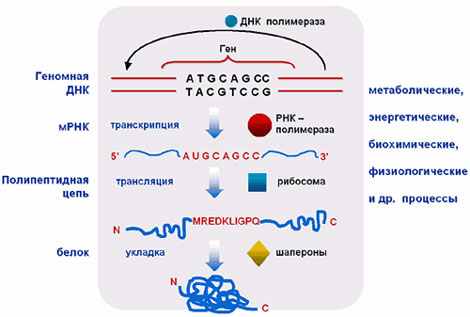
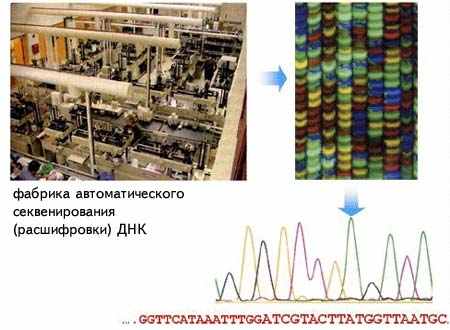
Суммарные объемы первичных экспериментальных данных только по молекулярно-генетическому уровню организации жизни превышают сотни терробайт. В результате автоматической расшифровки нуклеотидных последовательностей в молекулярной биологии и генетике за последние 20 лет произошел информационный взрыв. Объемы получаемых данных поражают воображение.
2.1.1Автоматическая расшифровка нуклеотидных последовательностей
2.1.2Характеристика генома человека
Н
 апример, длина генома человека составляет более 6 миллиардов пар оснований и он содержит более 30 тысяч генов. При его расшифровке получены данные объемом десятки терробайт о физических и цитогенетических картах хромосом, их нуклеотидных последовательностях, локализации генов, мутациях: выявлено не менее 1.5 миллиона мутаций, по которым геномы людей отличаются друг от друга.
апример, длина генома человека составляет более 6 миллиардов пар оснований и он содержит более 30 тысяч генов. При его расшифровке получены данные объемом десятки терробайт о физических и цитогенетических картах хромосом, их нуклеотидных последовательностях, локализации генов, мутациях: выявлено не менее 1.5 миллиона мутаций, по которым геномы людей отличаются друг от друга. 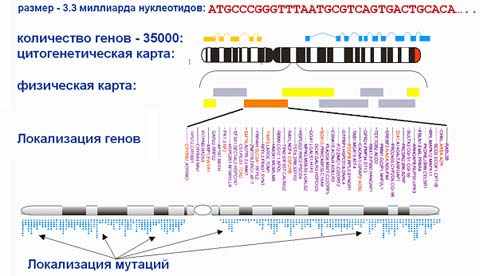
2.1.3
2.1.4Экспериментальные данные
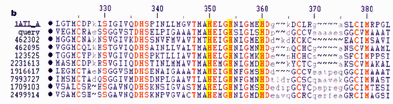
Расшифрованы структуры геномной ДНК тысяч вирусов, десятков бактерий, геномы дрожжей, дрозофилы, ряда животных и растений.
Расшифрованы аминокислотные последовательности миллионов белков и более 15 тысяч пространственных структур белков. Технология ДНК-чипов позволяет количественно измерять экспрессию десятков тысяч генов одновременно в отдельной клетке. Разворачиваются исследования по протеомике, направленные на расшифровку первичной и пространственной структур всех белков человека и бактерий (миллионы молекул). Огромные экспериментальные данные накапливаются при изучении разнообразия геномов человека и животных.
В связи с актуальностью данной темы, целью настоящей работы явилось показать способы применения достижений информационных технологий при исследовании в области молекулярной генетики.
2.2 Глава 1 (способы применения IT в молекулярной генетике.).
В связи со спецификой молекулярной генетики как отрасли генетических знаний способы применения достижений информационных технологий различны. Условно их можно разделить на две группы. К первой относится использование различного рода специальных поисковых систем (PubMed, NCBI и др.) Ко второй группе принадлежат прикладные программы, которыми пользуется исследователь при своих экспериментах (BLAST, OligoCalc, и др.)
2.2.1 Применение IT для поиска необходимой информации.
Для поиска нужной информации о той или иной нуклеотидной последовательности, организме-объекте или научной статьи по заданной тематике, автору возможно использование различных поисковых систем. Самым простейшим методом является поиск информации через Google. Принцип работы данной системы достаточно известен, поэтому не буду подробно на нем останавливаться. Для поиска более подробной информации существует ряд специальных поисковых систем и баз данных. Перечислю некоторые из них:
- MedHunt(ссылка скрыта). MedHunt использует людей и поисковые технологии, чтобы формировать свою индексную базу. Поиск может быть ограничен регионом. Также доступен французский интерфейс.
- Biocrawler(ссылка скрыта). Каталог и поисковый сервер для поиска информации по биологии.
- Medical World Search(ссылка скрыта). Medical World Search разработан, чтобы улучшить доступ к медицинской информации профессиональным медикам и потребителям. Medical World Search сосредоточен на развитии интеллектуального поискового сервера используя недавние усовершенствования в медицинской информатике в отличие от программ, которые ориентируются на простом поиске слова.
- NCBI(ссылка скрыта). Учреждённый в 1998 году ресурс по молекулярной биологии. NCBI создаёт базы данных, проводит исследования, разрабатывает программные средства, распространяет биомедицинскую информацию – всё для лучшего понимания процессов на молекулярном уровне, затрагивающих здоровье и болезни.
- Echidna Medical Search(ссылка скрыта). Поиск медицинской информации на австралийских медицинских сайтах.
- Galenicom(ссылка скрыта). Поисковый сервер, осуществляющий поиск на испанских медицинских ресурсах.
- Medisearch(ссылка скрыта). Интернациональные медицинские ресурсы.
- MedNets(ts.com)MedNets – каталог ресурсов медицинской тематики. Содержит более 20000 ссылок на лучшие медицинские ресурсы.
Наиболее часто используемым способом поиска информации о научных статьях в области молекулярной биологии, генетики, микробиологии, медицине является сайт национальной медицинской библиотеки США – PubMed(ссылка скрыта). Для удобства тематического поиска и анализа биомедицинской информации, все журнальные статьи в Index Medicus и Medline проиндексированы по определенным ключевым словам или терминам, которые включены в специальный словарь под названием “Medical Subject Headings” (MESH). Использование такого подхода обеспечивает однообразие и преемственность в иерархической структуризации биомедицинской литературы. Термины MESH и древовидная структура их взаимоотношений пересматриваются ежегодно.
Поиск информации в Pubmed достаточно прост, но для более эффективной работы необходимо знать некоторые особенности его организации и принципы функционирования. Домашняя страница сайта www.pubmed.gov содержит, прежде всего, поле для выражения запроса, которое находится в верхней части экрана. Сразу под ним находится строка со ссылками на дополнительные настройки и инструменты, позволяющие улучшить стратегию поиска. Слева находится боковая колонка со ссылками на помощь по поиску, самоучитель и различные службы PubMed. В поле запроса можно ввести любую комбинацию нужных терминов. Для выполнения самого поиска следует кликнуть указателем мыши на кнопку “GO”.
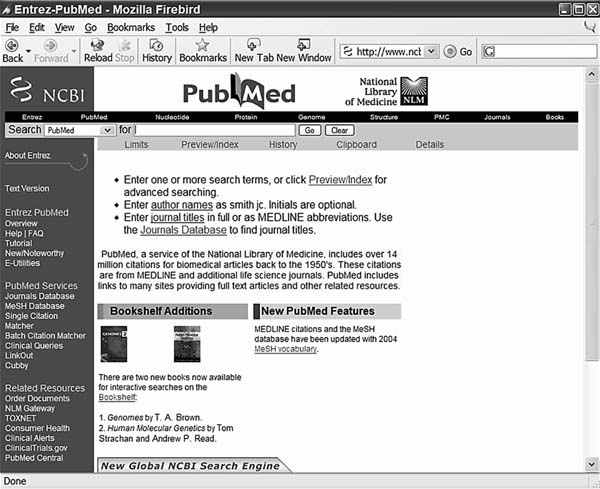
По умолчанию, поисковая система PubMed автоматически производит картирование терминов введенных Вами в строку выражения для поиска. Картирование заключается в том, что программа ищет соответствие между терминами введенными пользователем, и терминами, которые являются стандартными и содержатся в нескольких справочниках или словарях PubMed. Поиск и сравнение происходит последовательно по четырем справочникам. Прежде всего – это словарь MESH, затем словарь названий журналов, затем словарь фраз и уже затем – индекс авторов.
Словарь MESH основан на языке UMLS (Единая система медицинского языка), который имеет синонимы или лексические варианты английского языка для стандартных медицинских терминов. В результате картирования, в окончательное выражение для поиска попадают не только введенные Вами термины, но и их синонимы и стандартные термины, а также термины, описывающие более частные детали, которые в древовидной структуре MESH ответвляются от заданного термина. Окно просмотра словаря MeSH (доступно через ссылку в левой части экрана) можно использовать для того, чтобы найти нужные Вам термины. Для того, чтобы просмотреть термин в окне просмотра MESH, введите его в поле для выражения поиска и кликните кнопку “Go”(Пуск). Если термина, или его синонимов нет в словаре, на экран будет выведен список возможных терминов. Если Ваш термин, или связанный с ним синоним, имеется в словаре, то Вам будут представлены следующие опции:
- определение термина,
- кнопка “Add” для добавления этого термина в поле выражения для поиска,
- древовидная структура терминов с выделением жирным шрифтом Вашего термина,
- список подразделов термина (например, осложнения, лекарственная терапия, хирургическое лечение),
- опция ограничения термина только основной темой,
- опция для выключения развертывания термина.
Базу данных журналов можно просмотреть путем выбора ссылки из меню находящегося в левой части экрана. При помощи этой ссылки Вы можете выбрать нужный журнал и рефераты в нем. Журнал можно найти по следующим признакам: полному названию журнала, международному стандартному серийному номеру (ISSN), сокращению названия, принятому в Medline, уникальному идентификационному номеру национальной медицинской библиотеки США – NLM ID. Для того, чтобы получить список журналов, имеющих полные тексты статей, доступные через Интернет, нужно выбрать ссылку "journals with links to publisher Web sites", которая находится в нижней части экрана. Этот список постоянно обновляется за счет новых журналов. Однако, следует иметь ввиду, что многие журналы требуют для доступа к статьям регистрацию, подписку и оплату. Краткое стандартное описание журнала в базе данных включает название журнала, сокращение, принятое в MEDLINE, NLM ID, сокращение принятое международной организацией стандартизации (ISO), ISSN, и меню ссылок.
Если при поиске слово не найдено ни в словаре MESH, ни в словаре названий журналов и ни в словаре фраз, то поиск ведется в словаре авторов в том случае, если после него стоят отдельно одна или две буквы. Если слово не будет найдено и в этом словаре, то поиск этого слова будет вестись по всем полям базы данных MedLine. Если введенная фраза не распознается программой PubMed должным образом, а слова фразы разделяются на отдельные термины, имеет смысл заключить ее в двойные кавычки. Это связано с тем, что поиск отдельных слов фразы в тексте, скорее всего, приведет к выводу ссылок на большое количество статей, не имеющих отношения к предмету поиска, тогда, как поиск фразы заключенной в кавычках будет осуществляться без изменения в индексе фраз всех статей PubMed. Если такая фраза действительно содержится в базе данных PubMed, результаты поиска будут значительно более точными. Если фраза не найдена ни в списке фраз, ни в индексе, то заданные кавычки просто игнорируются. Следует иметь в виду, что при использовании кавычек не происходит картирование терминов, которое во многих ситуациях может быть очень полезным. В связи с этим, иметь смысл попробовать провести поиск нужной информации с термином с использованием кавычек и без кавычек.
Если нужно найти публикации определенного автора, но известна только его фамилия, следует обязательно поместить после нее метку [au], что означает, что данное слово следует искать в поле фамилии автора. Если известен хотя бы первый инициал, то его указание после фамилии автора достаточно для того, чтобы программа искала эту фамилию в нужном поле. Также можно задавать для выражения поиска усеченные слова (truncate) с добавлением знака звездочки (*) с тем, чтобы поиск происходил по всем словам содержащим один корень. Такой метод поиска часто именуется также как “wildcard”. Однако, и здесь следует помнить, что введение слова в такой форме также отключает автоматическое картирование фраз. Знак замещения одной буквы (?), принятый в некоторых других поисковых системах, в PubMed не используется.
При задании выражения для поиска можно использовать слова булевской логики (операторы), которые определяют взаимоотношения между фразами или терминами. Используют оператор AND (“И”), для отбора информации, которая содержит каждую из фраз, объединенных этим оператором. Этот оператор не накладывает условий на местоположение фраз, необходимо только, чтобы фразы находились где-нибудь в одном и том же документе. Используют оператор OR (“ИЛИ”) для отбора информации, которая содержит, по крайней мере, одну из фраз, объединенных этим оператором. Используют оператор NOT (“НЕ”), для отбора информации не содержащей фразы, перед которой поставлен этот оператор. Операторы должны быть всегда введены в верхнем регистре (заглавными буквами). Программа читает булевские выражения слева направо. Для смены последовательности действия операторов можно использовать скобки. Фразы, заключенные в скобки, оцениваются в первую очередь и рассматриваются как единое целое.
Используют предварительный просмотр (Preview / Index – опция в строке под полем для введения выражения запроса) для того, чтобы просмотреть только количество позиций отобранных в результате выполнения поиска, отработки стратегии поиска путем включения нескольких терминов одновременно, добавления терминов, которые должны искаться в определенных полях, выбора терминов из Индекса.
Отобранные статьи могут быть выведены на экран в нескольких форматах. На практике, наиболее часто используются следующие форматы: Citation (Цитирование), Abstract (Реферат), Brief (Краткий). Все они помимо названий и указания источника статьи, содержат ссылки на полные тексты статей (если они доступны через Интернет), ссылки на схожие по теме статьи, книги и базы данных. Форматы Citation и Abstract содержат рефераты статей, если они имеются в первоисточнике.
PubMed содержит ссылки на статьи опубликованные начиная с 1966 года. Существует два типа дат: Entrez Date – дата, когда ссылка была добавлена в PubMed. Можно задать ограничение на эту дату блочно в интервале от 30 дней до 10 лет. Publication Date - дата, когда статья была опубликована. Можно использовать поля "From" (начиная с …) и "To" (заканчивая по …) для указания интервала допустимых дат публикации.
Часто за поиском небходимой информации, в частности методик, данных об интересующем организме-объекте исследования, векторах, ферментах, наборах для проведения реакций, составах сред и компонентах той или иной реакции обрашаются к рускоязычным сайтам, таким как ссылка скрыта и ссылка скрыта. Сайт "molbiol.ru" - нейтральная русскоязычная территория для тех, кто связан с биологией или молекулярной биологией. Цель проекта - создать в интернете известное всем "профсоюзное место встречи". Организаторы проекта считают, что их задача только подготовить и обустроить информационную площадку, которая будет наполняться и поддерживаться всем русскоязычным биологическим сообществом. Материалы, размещённые на сайте сохраняют исходное авторство. Сайт molbiol.ru организован Алексеем Солдатовым и Татьяной Бородиной Max-Planck-Institut fur Molekulare Genetik (Берлин, лаборатория H.Lehrach). В 1999-2001г проект финансировался Российским Фондом Фундаментальных Исследований (РФФИ). В 2003-2005г года расходы по содержанию сайта оплачивались за счёт пожертвований посетителей. С 2005г. проект содержится за счёт размещения на нём рекламных материалов. У специалистов в области молекулярной биологии и генетики есть возможность ссылаться на материалы сайта. На сайте можно (и нужно) публиковать всё, что интересно коллегам-биологам, специалистам в области молекулярной биологии и генетики:
- общую интересную и доступную информацию — в колонке новостей на главной странице;
- справочную информацию;
- методики;
- дополнения или поправки к методикам или обзорам;
- можно завести собственный раздел или собственное отделение Форума;
- объявления о поиске работы / сотрудника / коллаботатора;
- объявления о школах, конференциях, семинарах;
- обзоры по темам, связанным с биологией;
Что касается авторства, то вся информация сохраняет исходное авторство и имеет гиперссылку на автора (по желанию mail, почтовый или web-адрес). Всё можно прислать по электронной почте по адресу redactor@molbiol.ru как прикреплённый файл (с материалом в текстовом, RTF или MS Word формате). Возможно также создание статьи в режиме online. Создают тему в каком либо форуме и пишут в ней свою статью (не обязательно одним блоком; можно вставлять файлы и рисунки). После связываются с модератором форума или администратором сайта, они могут превратить тему в постоянную страницу сайта. Если пользователь зарегистрирован на форуме, то в результате получит полный контроль как над темой форума, так и над материалом постоянной страницы и сможет их модифицировать в любое удобное время.
На сайте присутствует большое количество справочной информации. Здесь исследователь может найти необходимые методики для проведения различного рода реакций. В протоколе методик присутствуют: 1) название; 2) прописи используемых растворов (если они выходят за рамки общеизвестных); 3) краткое описание преимуществ данного протокола; 4) в комментариях — обычные выходы продукта и затраты времени; 5) разделён собственно протокол и комментарии к нему (это позволит использовать протокол "за столом" непосредственно в ходе эксперимента). Для всех остальных материалов на сайте есть специальные формы:
- сообщение для главной страницы сайта;
- поиск работы (аспирантуры, дипломной или курсовой работы);
- поиск сотрудника (аспиранта, студента);
- поиск единомышленников или сотрудничества;
- работы и услуги (продам, подарю, выполню);
- работы и услуги (закажу, куплю);
- информация о компании.
Вторым русскоязычным ресурсом, пользующимся большим спросом у исследователей является сайт ссылка скрыта. Этот сайт является проектом фирмы Интерруссофт. Цель проекта - создать первый в России специализированный портал, посвященный биоинформатике и биотехнологии. Портал ведётся и развивается специалистами как в области биотехнологии, так и в области IT, и имеет специализированные разделы, ориентированные на:
- профессионалов;
- инвесторов и бизнесменов;
- всех интересующихся.
Сайт создан как русский аналог проекта ссылка скрыта открыт для сотрудничества всех, заинтересованных в развитии российских биотехнологий. В настоящее время портал rusbiotech.ru не является коммерческим проектом, однако он будет активно использоваться в различных научных и инвестиционных проектах.
Сайт может быть полезен для поиска необходимой информации о методиках, исследуемых организмах, реактивах, материалах и оборудовании. На сайте также содержится информация об организациях, проектах, грантах, а также научные публикации по геномике, молекулярной биологии, биоинформатике, биотехнологии.
2.2.2 Применение IT для проведения молекулярно-генетических исследований.
Часто для получения необходимой информации бывает не достаточно воспользоваться вышеуказанными поисковыми системами. Например, необходимо найти последовательность нуклеотидов определенного гена, подобрать праймеры к заданной нуклеотидной последовательности. Сравнение аминокислотных и нуклеотидных последовательностей является важным звеном исследований молекулярной генетике, которое позволяет идентифицировать семейства генов, относить к ним секвенированные последовательности, устанавливать их структурные и функциональные взаимоотношения. В настоящее время, когда секвенируются целые геномы, значение подобных исследований постоянно возрастает. В таких случаях используют специальные программы.
К программам, используемым для сравнения последовательностей с последующим определением их сходства, относятся: ALIGN, AMAS, BLAST, BLAT, CLUSTAL, DiAlign, FASTA, HI, HMMER, MAP, MGA, OWEN, PipMaker, MultiPipMaker, T-Coffee и др. Наиболее часто используются программы CLUSTAL и BLAST (Basic Local Alignment Search Tool, основное средство поиска, основанное на локальных выравниваниях).

BLAST (англ. Basic Local Alignment Search Tool) — семейство компьютерных программ, служащих для поиска гомологов белков или нуклеиновых кислот, для которых известна первичная структура (последовательность) или её фрагмент. Используя BLAST, исследователь может сравнить имеющуюся у него последовательность с последовательностями из базы данных и найти последовательности предполагаемых гомологов. Является важнейшим инструментом для молекулярных биологов, генетиков, биоинформатиков, систематиков. Первое поколение BLAST программ появилось в начале 90-х годов прошлого столетия. Второе поколение программ данной серии представлено двумя вариантами: WU-BLAST 2 (Washington University BLAST 2) и NCBI BLAST 2 (National Center for Biotechnology information BLAST 2). Эти программы являются самыми быстрыми (скорость поиска на порядок выше программы FASTP и других алгоритмов) и чувствительными (определяют даже незначительное сходство последовательностей). Программы серии BLAST продолжают модифицироваться, при этом скорость поиска у последних версий BLAST приблизительно в 3 раза выше скорости оригинала.
Семейство программ серии BLAST делится на 5 основных групп:
1. Нуклеотидные – предназначены для сравнения изучаемой нуклеотидной последовательности с базой данных секвенированных нуклеиновых кислот и их участков:
- megablast – быстрое сравнение с целью поиска высоко сходных последовательностей,
- dmegablast – быстрое сравнение с целью поиска дивергировавших последовательностей, обладающих незначительным сходством,
- blastn – медленное сравнение с целью поиска всех сходных последовательностей и др..
2. Белковые – предназначены для сравнения изучаемой аминокислотной последовательности белка с имеющейся базой данных белков и их участков.
- blastp – медленное сравнение с целью поиска всех сходных последовательностей,
- cdart – сравнение с целью поиска гомологичных белков по доменной архитектуре,
- rpsblast – сравнение с базой данных консервативных доменов,
- psi-blast – сравнение с целью поиска последовательностей, обладающих незначительным сходством,
2.5. phi-blast – поиск белков, содержащих определенный пользователем паттерн и др.
3. Транслирующие – способны транслировать нуклеотидные последовательности в аминокислотные:
3.1. blastx – переводит изучаемую нуклеотидную последовательность в кодируемые аминокислоты, а затем сравнивает ее с имеющейся базой данных аминокислотных последовательностей белков,
3.2. tblastn – изучаемая аминокислотная последовательность сравнивается с транслированными последовательностями базы данных секвенированных нуклеиновых кислот,
3.3. tblastx – переводит изучаемую нуклеотидную последовательность в аминокислотную, а затем сравнивает ее с транслированными последовательностями базы данных секвенированных нуклеиновых кислот.
4. Геномные – предназначены для сравнения изучаемой нуклеотидной последовательности с базой данных секвенированного генома какого-либо организма (человека, мыши и др.)
5. Специальные – прикладные программы, использующие BLAST:
5.1. bl2seq – сопоставление двух последовательностей по принципу локальных выравниваний,
5.2. VecScreen – определение сегментов нуклеотидной последовательности нуклеиновой кислоты, которые могут иметь векторное происхождение и др.
Все выравнивания принято делить на глобальные (последовательности сравниваются полностью) и локальные (сравниваются только определенные участки последовательностей). Программы серии BLAST производят локальные выравнивания, что связано с наличием в различных белках сходных доменов и паттернов. Кроме этого локальное выравнивание позволяет сравнить иРНК с геномной ДНК. В случае глобального выравнивания обнаруживается меньшее сходство последовательностей, особенно их доменов и паттернов.
После введения изучаемой нуклеотидной или аминокислотной последовательности (запрос) на одну из web-страниц BLAST, она вместе с другой входной информацией (база данных, размера «слова» (участка), значение величины E и др.) поступает на сервер. BLAST создает таблицу всех «слов» (в белке – это участок последовательностей, который по умолчанию состоит из трех аминокислот, а для нуклеиновых кислот из 11 нуклеотидов) и сходных «слов». Затем в базе данных проводится их поиск. Когда обнаруживается соответствие, то делается попытка продлить размеры «слова» (до 4 и более аминокислот и 12 и более нуклеотидов) сначала без гэпов (пробелов), а затем с их использованием. После максимального продления размеров всех возможных «слов» изучаемой последовательности, определяются выравнивания с максимальным количеством совпадений для каждой пары запрос – последовательность базы данных, и полученная информация фиксируется в структуре SeqAlign. Форматер, расположенный на сервере BLAST, использует информацию из SeqAlign и представляет ее различными способами (традиционным, графическим, в виде таблицы).
Для каждой обнаруженной в базе данных программами BLAST последовательности необходимо определить, насколько она сходна с изучаемой последовательностью (запрос) и значимо ли это сходство. Для этого BLAST вычисляет число битов и величину Е (expected value, E-value) для каждой пары последовательностей.
При определении сходства ключевым элементом является матрица замен, так как она определяет показатели сходства для любой возможной пары нуклеотидов или аминокислот. В большинстве программ серии BLAST используется матрица BLOSUM62 (Blocks Substitution matrix 62% identity, блоковая матрица замен с 62% идентичности) . Исключением являются blastn и megablast (программы, которые выполняют нуклеотид – нуклеотидные сравнения и не используют матрицы аминокислотных замен).
С помощью модифицированных алгоритмов Смита-Уотермана или Селлерса определяются все пары сегментов (продленные «слова»), которые нельзя увеличить, так как это приведет к уменьшению показателей сходства. Такие пары продленных «слов» называются парами сегментов с максимальным сходством (high-scoring segment pairs, HSP). В случае достаточно большой длины изучаемой последовательностей (m) и последовательности базы данных (n) показатели сходства HSP характеризуются двумя параметрами K (размера области поиска) и ? (системы подсчета). Эти показатели необходимо указывать при приведении показателей сходства изучаемой последовательности и последовательности базы данных (S).
Для сравнения показателей сходства различных выравниваний независимо от используемой матрицы, их необходимо преобразовать. Для получения преобразованного показателя сходства (числа битов, Sґ) используют формулу:
Sґ = (?S – ln K)/ln 2 (1).
Величина Sґ показывает, насколько сходны последовательности (чем больше число битов, тем больше сходство). Так как в формулу расчета Sґ заложены показатели К и ?, то нет необходимости указывать их при приведении значений Sґ. Величина E (Е-value), соответствующая показателю Sґ, показывает достоверность данного выравнивания (чем ниже значение E, тем достовернее выравнивание). Она определяется по формуле:
E = mn 2 – Sґ (2).
Программы BLAST преимущественно определяют значение E, а не P (вероятности наличия хотя бы одного HPS с показателем, превышающим или равным S). Но при E < 0,01 значения P и E почти идентичны.
Величина E определяется по формуле (2) при сравнении лишь двух аминокислотных или нуклеотидных последовательностей. Сравнение изучаемой последовательности длиной m с множеством последовательностей базы данных может основываться на двух положениях. Первое положение состоит в том, что все последовательности базы данных одинаково сходны с изучаемой. Это подразумевает, что значение E для выравнивания с короткой последовательностью, содержащейся в базе данных, следует приравнять со значением E для выравнивания с длинной последовательностью. Для вычисления значения E по базе данных необходимо умножить значение E, полученное при попарном сравнении, на число последовательностей в ней. Второе положение заключается в том, что изучаемая последовательность более сходна с короткими, а не с длинными последовательностями, потому что последние часто состоят из различных участков (многие белки состоят из доменов). Если предположить, что вероятность сходства пропорциональна длине последовательности, то попарное значение E для последовательности базы данных длиной n надо умножить на N/n, где N – общая длина аминокислот или нуклеотидов в базе данных. Программы BLAST преимущественно используют этот подход для вычисления значений E по базе данных.
Теоретически локальное выравнивание может начинаться с любой пары нуклеотидов или аминокислот выровненных последовательностей. Однако HPS, как правило, не начинаются близко к краю (началу или концу) последовательностей. Для коррекции такого краевого эффекта необходимо вычислять эффективную длину последовательностей. В случае последовательностей длиной более 200 остатков происходит нейтрализация краевого эффекта.
Рассмотренные выше показатели разрабатывались для не содержащих гэпов местных выравниваний. Однако в ходе последующих исследований было установлено, что эти показатели могут использоваться и для выравниваний, содержащих гэпы.
При подборе последовательностей праймеров для постановки ПЦР-анализа часто пользуются программой OligoСalc. Для анализа в окно браузера копируют смоделированную нуклеотидную последовательность, указывая 5’ и 3’-концы, затем нажимают на панель Calculate. В появившемся окне содержится вся информация о данном праймере:длинна, число Г-Ц пар, температура плавления. Для анализа самокомплементарности и наличия шпилечных структур в браузере необходимо нажать на панель Check Self-complementarity. В появившемся окне содержится информация о наличии и числе двунитевых участков и шпилек.
2.3 Глава 2 (Применение IT в исследованиях по подготовке магистерской диссертации).
Подготовку магистерской диссертации невозможно представить без использования достижений современных информационных технологий. Ряд программ используется как при непосредственном проведении эксперимента, так и при оформлении полученных результатов. Одной из задач магистерской диссертации является исследование генетической организации плазмид биодеградации нафталина группы Inc-P7. В частности необходимо исследовать область генов, ответственных за репликацию (RepA-OriV). Исследование предполагали проводить при помощи ПЦР-анализа со специфическими праймерами, с последующей рестрикцией продуктов амплификации. Для провеления реакции ПЦР одним из важнейших компонентов являются праймеры. Праймеры – это короткие нуклеотидные последовательности длинной 15-30 нуклеотидов, комплементарные определенным участкам амплифицируемой ДНК. Праймеры к ряду известных последовательностей генов продаются в готовом виде. Если же к данной последовательности праймеров не существует, то подбор их осуществляется «вручную» с использованием ряда компьютерных программ. Плазмиды, выделенные в лаборатории из природных штаммов являются главным объектом исследования. Для начала необходимо знать сиквенс исходной матричной последовательности необходимого гена. Праймеры будут построены на основании сиквенса последовательности плазмиды pND6-1. Выбор именно этой плазмиды обусловлен тем, что это единственная, на данный момент плазмида биодеградации нафталина, принадлежащая к группе Inc-P7, нуклеотидная последовательность которой определена и находится в геномной базе данных. Для поиска был использован ресурс ссылка скрыта. В строке браузера был задан поиск последовательности (Nucleotide) данной плазмиды. В появившемся окне содержится информация о каталожном номере данного сиквенса, информация об организме-хозяине, а также характеристика основных открытых рамок считывания. Возможно также вести поиск по геному (Genome) и бактериальному хозяину (Organism). Пользуясь полученной информацией находят номера необходимых рамок считывания, их координаты. Затем, согласно координатам выделяют нужную последовательность в общем сиквенсе и копируют ее в Word. Затем «вручную» подбирают прямой и обратный праймеры, использую правила комплементарности. Затем полученные последовательности проверяют с помощью программы OligoCalc. Если и прямой, и обратный праймер имеют одинаковую температуру плавления, Г-Ц индекс не более 50%, длинну не более 30 нуклеотидов, не содержат двунитевых участков и шпилек, то можно заказывать их изготовление. Готовые праймеры используют для постановки ПЦР-анализа.
2.4Глава 3 (Заключение).
Молекулярная генетика своими замечательными открытиями оказала плодотворное влияние на все биологические науки. Она явилась той основой, на которой выросла молекулярная биология, значительно ускорила прогресс биохимии, биофизики, цитологии, микробиологии, вирусологии, биологии развития, открыла новые подходы к пониманию происхождения жизни и эволюции органического мира. Достижения молекулярной генетики, внёсшие огромный теоретический вклад в общую биологию, широко использованы в практике сельского хозяйства и медицины (т. н. генная инженерия путём замены вредных генов полезными, в том числе искусственно синтезированными; управление мутационным процессом; борьба с вирусными болезнями и злокачественными опухолями путём вмешательства в процессы репликации нуклеиновых кислот и опухолеродных вирусов; управление развитием организмов посредством воздействия на генетические механизмы синтеза белка и т. д.). Перспективность практического применения достижений молекулярной генетики подтверждается успехами, достигнутыми на модельных объектах. Однако современность диктует свои правила игры, и молекулярная генетика в последнее время перестала быть в чистом виде практической. Без использования информационных технологий практически не возможно начать исследование. На огромном пути от поиска научных публикаций по заданной теме до поиска нужного гена в электронных геномных базах данных, постановки сложнейших аналитических реакций современные достижения IT являются неотъемлемой частью и большим подспорьем. Без использования специализированных компьютерных программ иногда не возможно проанализировать тот огромный объем информации, который получает специалист в области молекулярной генетики в ходе своих исследований. Ну и, наконец, сейчас ни одно оформление магистерской, кандидатской, докторской диссертации не обходится без применения IT. Программы Word, Excel, PowerPoint являются неотъемлемыми элементами, сопровождающими научный и околонаучный (конференции, симпозиумы, семинары) процесс. Огромное значение IT в современной науке очевидно, и в дальнейшем, возможно, что эти две области знаний будут и дальше развиваться параллельно и пополнять одна другую новой информацией.
2.5
2.6Список литературы к реферату.
2.7
3Интернет ресурсы в предметной области исследования.
- ссылка скрыта - нейтральная русскоязычная территория для тех, кто связан с биологией или молекулярной биологией. Цель проекта - создать в интернете известное всем "профсоюзное место встречи".У специалистов в области молекулярной биологии и генетики есть возможность ссылаться на материалы сайта. На сайте можно (и нужно) публиковать всё, что интересно коллегам-биологам, специалистам в области молекулярной биологии и генетики.
- ссылка скрыта - первый в России специализированный портал, посвященный биоинформатике и биотехнологии. Портал ведётся и развивается специалистами как в области биотехнологии, так и в области IT. Сайт может быть полезен для поиска необходимой информации о методиках, исследуемых организмах, реактивах, материалах и оборудовании. На сайте также содержится информация об организациях, проектах, грантах, а также научные публикации по геномике, молекулярной биологии, биоинформатике, биотехнологии
- ссылка скрыта - сайт национальной медицинской библиотеки США , для удобства тематического поиска и анализа биомедицинской информации, все журнальные статьи в Index Medicus и Medline проиндексированы по определенным ключевым словам или терминам, которые включены в специальный словарь под названием “Medical Subject Headings” (MESH). Использование такого подхода обеспечивает однообразие и преемственность в иерархической структуризации биомедицинской литературы.
- ссылка скрыта. - Учреждённый в 1998 году ресурс по молекулярной биологии. NCBI создаёт базы данных, проводит исследования, разрабатывает программные средства, распространяет биомедицинскую информацию – всё для лучшего понимания процессов на молекулярном уровне, затрагивающих здоровье и болезни.
- ссылка скрыта - Небольшая подборка бесплатного программного обеспечения – трёхмерное моделирование, программы для автоматизации анализа последовательностей нуклеиновых кислот и белков, моделирования кинетики реакций и др.
- ссылка скрыта - Программа, которая сравнивает нуклеотидные и аминокислотные последовательности в ДНК и белках; поиск отличий и эволюционного расстояния.
- ссылка скрыта. - представляет собой двухуровневый указатель ресурсов по основным разделам биологии и смежным областям.
- http://ссылка скрыта - собрана обширная коллекция адресов узлов Интернет, относящихся к различным областям знаний. Коллекция организована в соответствии с достаточно подробным рубрикатором. Разделы Virtual Library ведутся различными лицами и организациями на добровольной основе.
- ссылка скрыта. - При поиске в Интернет информации биологического характера весьма полезным может оказаться раздел ссылка скрыта британской информационной системы ссылка скрыта , содержащий весьма детальный перечень разделов наук в области биологии со ссылками на сервера Интернет, поддерживающие информацию по каждому из разделов.
- ссылка скрыта - представляет собой интернет-ресурс, предназначеенный в помощь профессионалам в области медико-биологических наук. Сайт содержит (по утверждению владельцев) более 35000 ссылок на различные медико-биологические ресурсы (журналы, базы данных, предметный указатель ссылок на указатели ресурсов по различным разделам биологии и медицины и т.п.).
4Действующий личный сайт в WWW (гиперссылка).
ссылка скрыта.
5Граф научных интересов.
магистрантки Черновой А. И. Биологический факультет.
Специальность биология
| Смежные специальности
| Основная специальность 03.00.15 – генетика Области исследований:
| Сопутствующие специальности
|
6Презентация магистерской (кандидатской) диссертации.
- Гиперссылка:
- Презентация:
Слайды презентации к защите магистерской диссертации см. в приложении.
7Список литературы к выпускной работе.
- ссылка скрыта.
- ссылка скрыта
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.
- ссылка скрыта.