Экспрессия гена и белка марганцевой супероксиддисмутазы в клетках с p53 (+/+) и p53 (-/-) геномом
| Вид материала | Диплом |
- Курс лекций 1999-2000, 11782.85kb.
- Николай Владимирович Максимович, Игорь Арсениевич Стогов, Андрей Эдуардович Фатеев,, 1978.73kb.
- Чистякова Елена Андреевна Открытый урок, 128.48kb.
- 1. 1 Природа генів, 282.45kb.
- Вопросы к экзамену по курсу «Молекулярная биология» (вечернее отделение), 36.64kb.
- Урок №94 тема. Основные этапы биосинтеза белка, 33.42kb.
- Методическая разработка урока по теме «Биосинтез белка», 59.93kb.
- Молекулярный уровень, 31.47kb.
- Всероссийская олимпиада школьников по обществознанию 2007/2008 гг. Региональный этап., 139.44kb.
- Лекция 2: Принцип Дирихле. Формулировка: Если в n "клетках", 166.11kb.
Таблица 1. Первичные радикалы, образующиеся в нашем организме
| Название радикала | Структура радикала | Ферментная система, ответственная за образование радикала | Биологическая роль радикала |
| Супероксид | ·OO- | НАДФН-оксидаза | Антимикробная защита |
| Нитроксид | ·NO | NO-синтаза | Фактор расслабления сосудов |
| Убихинол | ·Q | Дыхательная цепь митохондрий | Переносчик электронов |
Таблица 2. Вторичные радикалы
| Название радикала | Структура радикала | Образуется в реакции |
| Супер радикал гидроксила | •ОН | Fe²++HOOH = Fe³++ HO-+•ОН Fe²++ClO-+H+ = Fe³++Cl-+•OH |
| Липидные радикалы | LO• L• LOO• | Fe²++LOOH = Fe³++HO-+LO• LO•+LH = LOH+L• L•+O2 = LOO• |
Супероксидный анион-радикал (О2-)
Кроме полного четырёхэлектронного восстановления молекулы кислорода до воды в дыхательной цепи митохондрий происходит и одноэлектронное восстановление с образованием О2-, который при взаимодействии с протоном переходит в гидроперекисный радикал (НО2), представляющий собой слабую кислоту [Зенков, Меньщикова, 1993]. Так как анион О2-- имеет заряд, он плохо мигрирует через мембраны в отличии от НО2. О2- - относительно слабый окислитель и во многих биологических системах выступает в качестве донора электронов, восстанавливая ряд соединений. При окислительном фосфолирировании примерно 1% кислорода восстанавливается до супероксида. Основные источники его образования – ферментативные системы: НАД(Ф)Н-оксидаза фагоцитирующих клеток, ксантиноксидаза, митохондриальная цитохром с-оксидаза и микросомальные монооксигеназы [Комов В.П., 2004].
Пероксид водорода (Н2О2).
Присоединение двух электронов к молекуле кислорода или одного электрона к супероксид-аниону сопровождается образованием двухзарядного аниона О22-. В свободном состоянии такой анион не существует, так как энергия связывания атомов кислорода становится отрицательной. Присоединяя протоны, он переходит в Н2О2 или НО2-, при этом преобладает Н2О2. Пероксид водорода относят к окислителям средней силы. Он относительно стабилен и может мигрировать через клеточные мембраны и вступать в реакции с клеточными компонентами, достаточно удалёнными от места синтеза. В живых организмах источниками Н2О2 служат ферментативные реакции с оксидазами, переносящими два электрона на молекулу кислорода: ксантиноксидазой, оксидазой аминокислот, альдегиддегидрогеназой и др., а также реакция дисмутации, катализируемая супероксиддисмутазой. Пероксид служит источником возникновения ОН. Кроме того, в присутствии миелопероксидазы происходит образование высокореакционных гипогалоидов (HOCI, HOBr, HOI, HJSCN), которые участвуют в защитных реакциях (Зенков, Меньщикова, 1993).
Накопление Н2О2 представляет большую опасность для клеток, так как при одноэлектронном восстановлении Н2О2, происходящем в присутствии свободных ионов Fe2+ или Cu+ (реакция Фентона), образуется высокоактивный гидроксил ОН:
Н2О2 + Fe2+=ОН- + ОН+ Fe3+
Гидроксильный радикал (ОН).
Этот короткоживущий свободный радикал является наиболее реакционноспособным, он может разрывать любую С-С или С-Н связь. Основным источником служит реакция Фентона. Обратное восстановление Fe3+ возможно в реакции с О2-, а также при взаимодействии с аскорбиновой кислотой, глутатионом, цистеином и другими легко окисляющимися органическими соединениями. ОН участвует в реакциях фагоцитоза, оказывает повреждающее действие на многие клеточные структуры, индуцирует образование органических радикалов [Турпаев, 2002].
Для защиты клеток от образования ОН большое значение имеют иммобилизация ионов железа и удаление лабильных гемовых групп [Овчинников Ю.А., 1987]. В этих процессах принимают участие соединения, способные хелатировать ионы: ферредоксин, металлотионеин и пиколиновая кислота (продукт расщепления триптофана), а также расщепляющая гемовое кольцо оксигеназа. Оксид азота может образовывать с ионами железа нитрозильные комплексы. Низкомолекулярные, легко окисляющиеся соединения, такие, как урацил, мочевая кислота, салицилаты, глюкоза, диметилсульфоксид, ингибируют радикалы ОН при очень высоких концентрациях.
Синглетный кислород (О2-)
Изменение спина одного из электронов, находящихся на π-орбиталях в молекуле кислорода, приводит к образованию возбуждённого синглетного состояния, энергия которого выше энергии основного триплетного состояния. Высокая реакционная способность приводит к тому, что он легко вступает в окислительные реакции с органическими соединениями, принимает участие в инициировании перекисного окисления липидов. В живых организмах не выявлено специальных систем генерации синглетного кислорода, но во многих ферментативных реакциях с участием супероксиддисмутазы, каталазы, пероксидазы отмечается его возникновение как сопутствуещего продукта [Зенков, Меньщикова, 1993].
1.2.2. Оксидативные повреждения клетки.
Реакция цепного окисления липидов играет исключительную роль в клеточной патологии. Реакциия протекает в несколько стадий, которые получили название «инициирование», «продолжение», «разветвление» и «обрыв цепи».Важно знать не только механизм пероксидации липидов, но и биологические последствия этого процесса.
Увеличенное образование свободных радикалов в организме и связанное с этим усиление процессов пероксидации липидов сопровождаются нарушениями в свойствах биологических мембран и функционировании клеток. Наиболее изучены три прямых следствия перекисного окисления липидов. Первое из них состоит в том, что перекисное окисление липидов сопровождается окислением тиоловых групп мембранных белков. Это может приводить в результате к неферментативной реакции SH-групп со свободными радикалами липидов. При этом образуются сульфгидрильные радикалы, которые затем взаимодействуют с образованием дисульфидов либо окисляются кислородом с образованием производных сульфоновой кислоты [Владимиров Ю.А., Азизова О.А., Деев А.И., 1992].
Второй результат перекисного окисления липидов связан с тем, что продукты пероксидации обладают способносстью непосредственно увеличивать ионную проницаемость липидного бислоя. Показано, что продукты перекисного окисления липидов делают липидную фазу мембран проницаемой для ионов водорода и кальция. Это приводит к потере митохондриями способности осуществлять синтез АТФ, и клетка оказывается в условиях энергетического голода. Одновременно в цитоплазму выходят ионы кальция, которые повреждают клеточные структуры [Владимиров Ю.А., 1987].
Последний и самый важный результат пероксидации – это уменьшение стабильности липидного слоя, что может привести к электрическому пробою мембраны собственным мембранным потенциалом. Электрический пробой приводит к полной потере мембранной её барьерных функций [Владимиров Ю.А., 1987].
1.2.3 Основные ферменты АОС.
Антиоксидантная система (АОС) – система противоокислительной защиты, противостоящая повреждающему действию свободных радикалов и активных форм кислорода [Казимирко В.И., 2004].
Система защиты от активных форм кислорода включает два остновных способа: неферментативный и ферментативный. Главным действующим звеном неферментативной защиты являются антиоксиданты – соединения, способные уменьшать интенсивность свободнорадикального окисления, нейтрализовать свободные радикалы путём обмена своего атома водорода (в большинстве случаев) на кислород свободных радикалов. Они могут быть природного (биооксиданты) и синтетического происхождения [Комов В.П., 2004].
Антиоксиданты имеют подвижный атом водорода и поэтому реагируют со свободными радикалами, а также с катализаторами свободнорадикального окисления и, прежде всего, с ионами металлов переменной валентности. Подвижность атома водорода обусловлена нестойкой связью с атомами углерода (С-Н) или серы (S-Н). По мнению ряда исследователей [Богач П.П. и соавт., 1981; Кучеренко Н.Е., Васильев А.Н., 1985], образующиеся свободные радикалы антиоксидантов малоактивны и выводятся из организма в виде молекулярных соеденений – продуктов взаимодействия с другими антиоксидантами (токоферолами, хинонами, витаминами группы К, серосодержащими соеденениями).
Ряд ферментов противостоит действию активных радикалов на клетку и эффективно обезвреживает эти соединения. Такой способ защиты от активных форм кислорода называется ферментативным [Комов В.П., 2004].
Первую линию защиты от свободных радикалов составляют такие антиоксидантные ферменты, как супероксиддисмутаза (СОД), каталаза и пероксидаза [Берберова Н.Т., 2000].
Супероксиддисмутазы
Супероксиддисмутазы – металлоферменты, катализирующие реакции дисмутации, в которых супероксид выступает одновременно и как окислитель, и как восстановитель:
2О2-→ Н2О2+О2
Супероксиддисмутазы находятся во всех клетках, потребляющих кислород. Скорость реакции чрезвычайно высока и лимитируется только скоростью диффузии О2-. Каталитический цикл этих ферментов включает восстановление и окисление иона металла на активном центре фермента. Существуют три изоформы супероксиддисмутазы [Комов В.П., 2004; Казимирко В.И., 2004; Fridovich I., 2001]. Медь-цинковая форма супероксиддисмутазы (CuZnSOD) локализована в основном в цитозоле и межмембранном пространстве митохондрий эукариотов, марганцевая форма (MnSOD) содержится в митохондриях и бактериях, а железосодержащая супероксиддисмутаза обнаружена у микроорганизмов [Поберезкина Н.Б., Осинская Л.Ф., 1989].
Супероксиддисмутаза осуществляет инактивацию радикалов кислорода, которые могут возникнуть в ходе биологических реакций переноса электронов или при воздействии металла с переменной валентностью, ионизирующего, УФ излучения, ультразвука, гипребарической оксигенации, различных заболеваний.
Каталаза.
Почти во всех животных клетках и органах определяется каталазная активность. Особенно богаты каталазой клетки печени, почек, эритроциты [Комов В.П., 2004]. Каталаза предотвращает накопление в клетке Н2О2, образующейся при аэробном окислении восстановленных флавопротеидов из О2-.
2Н2О2 →О2+2Н2О
Каталаза относится к числу ферментов с наиболее высоким числом оборотов, то есть она может разложить 44 000 молекул Н2О2 в секунду. Для расщепления большого количества Н2О2 требуется малое количество фермента. Как и в случае с супероксиддисмутазой, скорость реакции определяется диффузией и не требует энергии для активации [Комов В.П., 2004].
Каталаза преимущественно локализована в пероксисомах [Halliwel B., Gutteridge J.M.C., 1989], внеклеточно каталаза находится в незначительных концентрациях. Наибольшая активность каталазы в организме характерна для печени [Fridovich I., 2001].
Пероксидазы.
Н2О2 может дисмутировать при действии пероксидаз – ферментов, использующих в качестве донора водорода различные органические соединения, например полифенолы (условно обозначаемые R):
R(OH)2+ Н2О2→2Н2О+RO2
Пероксидазы содержатся в животных тканях (кровь, печень, почки), но особенно активны эти ферменты в тканях высших растений. Как каталаза, так и пероксидаза могут утилизировать органические гидроперекеси (например, гидроперекись этила, надуксусную кислоту). Полагают, что в животных тканях каталаза действует как пероксидаза [Казимирко В.И., 2004].
Глутатионпероксидаза.
В эритроцитах, печени, хрусталике глаза имеется глутатионпероксидаза (GPx), которая содержит селен и спецефично окисляет восстановленный глутатион. С помощью восстановленного глутатиона осуществляется детоксикация Н2О2 и гидроперекисей, которые образуются при реакции активных радикалов кислорода с ненасыщенными жирными кислотами мембраны эритроцитов:
Н2О2+2GSH→ GSSG+2Н2О, где
GSH-восстановленный глутатион;
GSSG- окисленный глутатион.
В пептидной цепи GPx имеется остаток селеноцистеина-аналога цистеина, в котором атом серы замещён атомом селена. Селеноцистеин входит в активный центр фермента. GPx может восстанавливать гидроперекиси свободных жирных кислот, гидроперекиси фосфолипидов, эстерифицированных жирных кислот [Halliwel B., Gutteridge J.M.C., 1989].Функцией фермента является поддержание активного состояния многих ферментов, самопроизвольное окисление которых приводит к образованию дисульфидной группы.
- Взаимодействие антиоксидантной системы и p53.
- Структура промотора марганцевой супероксиддисмутазы.
Ген sod экспрессирует фермент, который является основным ферментом антиоксидантной защиты в организме. Супероксиддисмутаза осуществляет инактивацию радикалов кислорода, которые могут возникнуть в ходе биологических реакций переноса электронов или при воздействии металла с переменной валентностью, ионизирующего, УФ излучения, ультразвука, гипребарической оксигенации, различных заболеваний.
Предшественник MnSOD (SOD2) синтезируется в виде полипептидной цепи, содержащей транзитный пептид (24 а/кислоты). Далее осуществляется его перенос в митохондрии и процессинг, в результате которого образуется зрелый белок (MW~24кДа). Зрелый белок образует гомотетрамер (MW~100кДа), каждая субъединица которого связывает по одному атому марганца.
Известно, что в эукариотических клетках начальным этапом синтеза мРНК является образование транскрипционного комплекса, который состоит из РНК-полимеразы II и базальных факторов транскрипции. Во многих генах домашнего хозяйства, с которым и относится sod, в промоторе отсутствует ТАТА и СААТ-блоки, но выявляется последовательность gggcgg или соответственно GC-блок. Сборка транскрипционного комплекса происходит именно на этом блоке [Георгиев Г.П., 1989].
Ген sod2 состоит из 5 экзонов. Промоторная область данного гена содержит сайты связывания таких факторов транскрипции, как SP1 и AP-2. Sp1 связывает GC-бокс и является активатором транскрипции, а AP-2 выполняет противоположную функцию. Второй интрон гена содержит энхансер. Энхансер - это цис-элемент, который увеличивает транскрипцию в разных ориентациях и независимо от того, куда бы его не поместили. Энхансерная область содержит содержит сайты связывания для транскрипционных факторов c/EBP и NF-kB, которые являются посредниками воспалительных сигналов.
- P53 зависимая регуляция гена супероксиддисмутазы.
Впервые влияние p53 на MnSOD показано в 2000 году [Pani G., 2000]. Работу проводили на фибробластах с p53 (+/+) геномом и p53 (-/-) геномом. Результаты анализа активности белка в геле показали, что в клетках с p53 (-/-) геномом активность белка MnSOD выше по сравнению с клетками, имеющими p53 (+/+) геном. Кроме того, при сверхэкспрессии р53 в клетках HeLa экспрессия гена sod2 и активность MnSOD снижалась. Поскольку сайта связывания для p53 в промоторе гена sod2 крысы не обнаружено, авторы предпологают, что p53 репрессирует ген sod2 опосредованно, ингибируя транскрипционный фактор Sp1.
Позже было исследовано взаимодействие между p53 и MnSOD [Drane P., Dravard A. Et all, 2001]. Работа проводилась на двух родственных клеточных линия MCF-7 и MCF/R-A1, экспрессирующих p53 дикого типа и мутантный, соответственно. Используя эту модель, авторы данной работы показали, что уровень мРНК sod2 и ферментативная активность MnSOD ниже в клетках дикого типа по сравнению с мутантными клетками. При облучении клеток дикого и мутантного типа активация гена sod2, индуцированная форболовыми эфирами, может быть эффективно репрессирована белком p53 дикого типа. Авторы предполагают, что это связано с тем, что p53 может быть включен в репрессию гена sod2.
Также в работе было показано, что промотор гена sod2 может вызвать репрессию транскрипции репортерного гена со стороны эндогенного или экзогенного p53 дикого типа, указывая, что p53-зависимая репрессия транскрипции sod2 имеет место на уровне промотора. Известно, что р53-зависимая репрессия осуществляется с помощью белок-белковых взаимодействий p53 с различными транскрипционными факторами,а также с помощью связывания p53 с определёнными последовательностями ДНК. Авторы связывают это с тем, р53- зависимая репрессия супероксиддисмутазы, приводя к накоплению АФК, является одним из механизмов р53-зависимого апоптоза.
В данной работе также показано, что сверхэкспрессия белка MnSOD ведёт к репрессии активности промотора p53. Известно, что транскрипционный фактор NF-kB, регулирующий ген р53, чувствителен к окислительно-восстановительному потенциалу. Однако, репрессия активности промотора p53 со стороны MnSOD не зависит от ингибирования NF-kB этим ферментом. Вероятно, MnSOD изменяет активность какого-то другого транскрипционного фактора, включённого в репрессию активности промотора р53.
В 2004 году были получены данные, противоречащие концепции р53-зависимого ингибирования гена sod2. На клетках лимфомы и на фибробластах с геномами р53(+/+) и р53(-/-) показано, что р53 не ингибирует, а активирует экспрессию гена sod2. Было установлено, что активация p53 приводит к повышению экспрессии MnSOD и глутатионпероксидазы, при этом экспрессия каталазы не изменяется.
Также, как и в предыдущей работе, авторы связывают эти эффекты с механизмами р53-зависимого апоптоза. В связи с недостатком каталазы, экспрессия которой остается неизменной, повышенная активность MnSOD приводит к накоплению в клетках перексида водорода, оксидативному стрессу и апоптозу. Противоречия между последней и предыдущими работами авторы объяснили различиями в клеточных линиях [Hussain S.P. et all, 2004].
Выше изложенные литературные данные позволяют сделать вывод о существовании нескольких p53-зависимых путей регуляции гена sod2, по-видимому, связанных с накоплением супероксид радикала или перексид водорода.
В нашей работе исследовалась экспрессия гена sod2 на аналогичной модельной системе, однако для активации р53 использовался актиномицин D в небольших наномолярных концентрациях. Подобный подход позволяет моделировать в клетках условия, близкие к естественным и избежать некоторые артефакты, связанные с суперэкспрессией, используемой в других работах. Исследования проводились на клетках аденокарциномы молочной железы MCF-7 и немелкоклеточной карциномы легкого H1299, обладающих, соответственно, p53 (+/+) и p53 (-/-) геномами. Сопоставление эффектов, наблюдаемых в этих двух типах клеток, позволяет судить о р53-зависимых реакциях, в том числе и о реакции гена sod2 на воздействия, стимулирующие опухолевый супрессор р53.
Глава 2. Материалы и методы исследования.
Клеточная культура. Эксперименты проводились на двух линиях клеток человека – аденокарциномы молочной железы MCF-7, экспрессирующие ген p53, и карциномы лёгкого H1299, не экспрессирующие ген p53. Клетки выращивались при 370С в атмосфере 5% СО2 в среде DMEM (), содержащей 10% сыворотку новорожденных телят (PAA Laboratories GmbH, Австрия).
Для активации р53 в клетках MCF-7 в инкубационную среду добавляли актиномицин D (ActD) до конечной концентрации 5 nM. Для изучения р53-независимых реакций подобную процедуру параллельно проводили на клетках H1299.
Для исследования экспрессии белка р53 клетки H1299 и MCF-7 рассеивали в пластиковые 6-луночные планшеты в 3 мл среды DMEM. Через 48 ч после рассева клеток проводили обработку клеток актиномицином D (5 nM). Через 0 ч (контроль), 2, 4 и 8 ч после внесения ActD клетки промывали буфером PBS (1.47 mM NaCl, 2.68 mM KCl , 4.29 mM Na2HPO4, 1.47 mM KH2PO4) и лизировали в буфере, содержащем 500 mM HEPES, 150 mM NaCl, 1 mM EDTA, 25 mM NaF, 10 мкМ ZnCl2, 10% глицерин, 1% Triton X100, 1 mM DTT, 1 mM ФМС. Затем проводили денатурирующий электрофорез в полиакриламидном геле (ПААГ) и реакцию иммуноблотинга.
Денатурирующий электофорез белков. Белки в полученных белковых препаратах разделяли в вертикальном 10%-ном ПААГ, содержащем 0.1% додецилсульфат Na (SDS). Для электрофореза клеточные лизаты смешивали с 2-кратным буфером Лэмли в пропорции 1:1 и инкубировали в течение 5 мин при температуре +95оС []. На каждую дорожку наносили 20 мкг белков клеточного лизата. Электрофорез проводили в трис-глициновом буфере (25 mM трис-HCl, рН 8.3 и 191.8 mM глицин), содержащем 0.1% SDS в приборе для вертикального электрофореза VE-2 (Хеликон, Россия) при силе тока 20 mA. Для определения молекулярной массы белков использовался маркер для электрофореза белков (Хеликон, Россия).
Иммуноблотинг. Гель после электрофореза пропитывали буфером TB (47.9 mМ трис-HCl, 38.6 mM глицин, 10% SDS, 20% метанола) в течение 15 мин. Затем готовили "сэндвич" для переноса: гель укладывался на 2 листа фильтровальной бумаги, смоченной в TB, на гель укладывали лист нейлоновой мембраны Hybond C (Amersham), смоченный в TB, а сверху – еще 2 листа фильтровальной бумаги, смоченной в TB. "Сэндвич" помещали в аппарат для переноса, заполненный буфером TB, гель находился со стороны анода, а мембрана – со стороны анода. Перенос в буфере TB проводили при 100 mА 16 часов при +40С, с постоянным перемешиванием на магнитной мешалке.
После переноса мембрану инкубировали в течении 1 часа в блокирующем буфере - 5% бычий сывороточный альбумин (V фракция), 0.02% азид натрия на растворе TBST (100 mM трис-HCl pH 7.5, 150 mM NaCl, 0.05% Tween). Затем в течение 1 часа мембрану инкубировали в растворе первичных антител к р53 (Santa Cruz, США) в блокирующем буфере в разведении 1:1000. После отмывки в TBST (3 раз по 10 мин при комнатной температуре) мембрану инкубировали в растворе вторичных антител к иммуноглобулину G кролика, связанных с пероксидазой хрена (Amersham, США) в блокирующем буфере без азида натрия в разведении 1:5000. После отмывки в TBST (3 раз по 10 мин при комнатной температуре) проводили реакцию хемилюминесценции: мембрану инкубировали в течении 1 мин в растворе, содержащем 0.68 mM паракумаровой кислоты, 1.25 mM люминола, 0.1 М трис-HCl, pH8.5, 0.01% H2O2, а затем проводили автографию на рентгеновской плёнке Biomax MS-1 (Kodak, США).
Для исследования экспрессии генов супероксид дисмутазы 2 (sod2) и глицеральдегид-3-фосфат дегидрогеназы (gapdh) клетки H1299 и MCF-7 рассеивали в пластиковые чашки Петри диаметром 5.5 см в 5 мл среды DMEM. Через 48 ч после рассева клеток проводили обработку клеток актиномицином D (5 nM). Через 0 ч (контроль), 2, 4 и 8 ч после внесения ActD проводили очистку РНК.
Очистка РНК. Все растворы для работы с РНК готовили на воде, обработанной диэтилпирокарбонатом в течение 16 ч при +37оС с последующим автоклавированием при давлении 1 атм в течение 1 ч.
Для очистки РНК клетки лизировались на чашках добавлением 1 мл тризола (), лизат инкубировали при комнатной температуре 10 минут, затем в него добавляли 200 мкл смеси хлороформ/изоамиловый спирт (в соотношении 24:1), тщательно перемешивали в течение 15 с и после выдерживания при комнатной температуре в течение 3 мин пробы центрифугировали при 5500 g в течение 20 мин при +40С. Верхнюю фазу отбирали, РНК осаждали добавлением равного объема изопропилового спирта с последующим инкубированием при комнатной температуре 10 мин и центрифугированием при 10000 g в течение 10 мин при +40С. Супернатант сливали, осадок промывали 1 мл 75% этилового спирта с последующим центрифугированием при 10000 g в течение 5 мин при +40С. Осадок подсушивали и растворяли в растворе ТЕ pH 7.5 (10 mM трис-HCl, 10 mM ЭДТА). Количество РНК в пробах определяли спектрофотометрически по поглощению света при длине волны 260 нм (А260). Чистоту РНК контролировали по поглощению света при длине волны 230 нм (А230) и 280 нм (А280). Отношение А260/А230 во всех пробах составляло 1.8-2.0, а А260/А280 – 2.1-2.2
Обратная транскрипция с полимеразной цепной реакцией (ОТ-ПЦР). ОТ-ПЦР анализ sod2 и gapdh проводили по методике Мелендеза и Дэвиса [J. B.C., N31, vol. 271, 1996, 18898-18903] с небольшими модификациями.
Праймеры для амплификации (ЗАО Евроген, Россия) имели следующую последовательность:
5’- SOD2: 5’-TCCCCGACCTGCCCTACGAC-3’;
3’-SOD2: 5’-CATTCTCCCAGTTGATTACAT-3’;
5’-GAPDH: 5’-CATCATCCCTGCCTCTACTGG-3’;
3’-GAPDH: 5’-TCTCTTCCTCTTGTGCTCTTG-3’.
Для отжига праймеров на РНК 2 мкг полученной РНК растворяли в 10 мкл воды и добавляли 3'-праймеры (0.05 мкг/мл). Смесь инкубировали в течение 5 мин при температуре +95оС и медленно снижали температуру до +67оС. Затем пробы инкубировались в течение 30 мин при температуре +60оС. Для обратной транскрипции в пробы добавляли смесь дезоксинуклеозид-трифосфатов до конечной концентрации 0.05 mM (в расчете на нуклеотид каждого вида), буфер для обратной транскриптазы M-Mlv (ЦНИИ Эпидемиологии МЗ РФ, Россия) и 200 единиц обратной транскриптазы M-Mlv (ЦНИИ Эпидемиологии МЗ РФ, Россия). Синтез кДНК проводился при инкубировании проб в течение 1 ч при температуре +42оС, реакцию останавливали прогреванием проб при +95оС в течение 5 мин.
Амплификацию ДНК проводили в смеси, содержащей 5мкл синтезированной кДНК, 1 мкМ 5'-праймера, 1 мкМ 3'-праймера, 2.5 mM дезоксинуклеозид-трифосфатов, буфер для ДНК-полимеразы Taq (СибЭнзим, Россия) и 1.25 единицы ДНК-полимеразы Taq (СибЭнзим, Россия). Амплификацию проводили в программируемом термостате (Биоком, Россия) по следующей программе:
3 цикла
+94оС 1 мин
+60оС 2 мин
+72оС 2 мин
27 циклов
+94оС 15 с
+60оС 30 с
+72оС 15 с
1 цикл
+72оС 3 мин
Электрофорез ДНК, полученной в реакции ОТ-ПЦР, проводили в горизонтальных агарозных гелях с концентрацией агарозы 2%. Агарозу готовили на буфере ТАЕ (40 mМ трис- HCl, pH 8.0, 1 mM ЭДТА, 2.4 mM уксусная кислота). Электрофорез проводили в том же буфере в течение 1.5 ч при напряжённости электрического поля 1 В/см [Маниатис Т., Фрич Э., Сэмбрук Дж., 1984]. Для определения молекулярного веса фрагментов ДНК использовали маркер длин фрагментов ДНК "100bp+1.5kb" (СибЭнзим, Россия). Для окраски ДНК гели вымачивали в растворе бромистого этидия (0.5 мкг/мл) в течение 1 ч.
Для исследования активности клеточных супероксид-дисмутаз клетки H1299 и MCF-7 рассеивали в пластиковые 6-луночные планшеты в 3 мл среды DMEM. Через 48 ч после рассева клеток проводили обработку клеток актиномицином D (5 nM). Через 0 ч (контроль), 2, 4 и 8 ч после внесения ActD клетки промывали буфером PBS, а затем собирали резиновым скребком в PBS, содержащем 1 mM ФМС, 0.2 mM ЭДТА, 20 mM NH4Cl. Клетки разрушали в гомогенизаторе Даунса на льду. Гомогенат центрифугировали при 10000 g в течение 15 мин при +4оС, супернатант отбирали для определения активности SOD в геле.
Определение активности супероксид-дисмутаз в геле (зимограммы супероксид-дисмутаз). Белки в полученных белковых препаратах разделяли в вертикальном 12%-ном неденатурирующем ПААГ, аналогично денатурирующему электрофорезу, но без добавления SDS. В белковые препараты добавляли буфер Лэмли без SDS и β-меркаптоэтанола. Электрофорез проводили в трис-глициновом буфере без SDS в приборе для вертикального электрофореза VE-2 (Хеликон, Россия) в течении 12 часов при силе тока 20 mA.
После электрофореза гель окрашивали в растворе, содержащем 0.25 mM NBT, 1 mМ ЭДТА, 50 mM калий-фосфатный буфер, pH 8.0, в течении 45 мин в темноте. Далее гель промывали деионизованной водой и выдерживали при дневном свете до появления окраски геля, при этом зоны геля, содержащие белки SOD, остаются неокрашенными.
Цифровая обработка данных. Окрашенные агарозные гели с ДНК снимали на цифровую камеру GelImager (Хеликон, Россия) ерез красный светофильтр при освещении ультрафиолетовым светом (длина волны 256 нм). Зимограммы SOD и рентгеновские автографы сканировали на сканере ArcusII (Agfa, США). Цифровая обработка данных проводилась с помощью программы GelAnalysis (Хеликон, Россия).
Глава 3. Результаты исследования и обсуждение.
3.1.Активация экспрессии p53 в MCF-7.
Белок p53 активируется в ответ на различные типы стресса, включая повреждение ДНК химическими соединениями и физическими факторами, активацию онкогенов, гипоксию, гипертермию и др. [Комарова Е.А., Гудкова А.В., 2000].
В наших экспериментах белок p53 активировали с помощью актиномицина D (ActD), который является ингибитором транскрипции [Диксон М., Уэбб Э., 1982]. Актиномицин D является интеркалятором, он встраивается между парами азотистых оснований (G-C) в ДНК и блокирует работу РНК-полимеразы [Passarge E., 2001]. Механизм активации p53 заключается в том, что при изменении конформационной структуры ДНК свойства РНК-полимеразы изменяется. Белок BRCA I, являющийся сенсором повреждений, теряет связь с РНК-полимеразой и способствует активации p53 [Чумаков, 2004].
Эксперименты проводили на двух клеточных линиях: MCF-7 и H1299. Для того, чтобы выявить особенности экспрессии гена sod2 в клетках с p53-защитной системой (MCF-7) и в клетках не имеющих p53-зависимую систему (H1299), сравнивалась экспрессия гена sod2 при действии актиномицина D в этих клеточных культурах.
На рис.1 показана экспрессия белка p53 в клетках MCF-7 при действии актиномицина D. Из рисунка видно, что экспрессия белка p53 в контроле (до нанесения актиномицина D) низкая. После нанесения актиномицина D количество белка p53 постепенно увеличивается и достигает максимального значения к 8 часам, а затем, как видно из рисунка, постепенно понижается.
Таким образом, была показана активация белка p53 в клетках MCF-7 при добавлении ActD. В последующих экспериментах изучалось влияние p53 на

Рис.1 Результаты иммуноблотинга клеточных лизатов MCF-7 c антителами к p53.
К- лизированные клетки MCF-7 lдо нанесения ActD;
4, 6, 8, 10, 12- лизированные клетки MCF-7 через 4 ч, 6 ч, 8 ч, 10 ч и 12 ч после нанесения ActD соответственно.
различных этапах действия актиномицина D, то есть в различных концентрациях, на экспрессию гена антиоксидантной системы – супероксиддисмутазы.
3.2. Изменение экспрессия гена sod2 в клетках MCF-7 и H1299 при действии актиномицина D.
Согласно данным литературы сверхэкспрессия гена p53 способствует репрессии гена sod2, и эта репрессия происходит на уровне промотора. Авторы связывают это с тем, что p53 стимулирует апоптоз путём сложной реакции, включающей репрессию транскрипции ряда генов, среди которых некоторые кодируют белки с антиапоптозной активностью: bcl-2, MAP4 и супероксиддисмутаза, связанная с окислительно-восстановительным потенциалом и играющих роль в продукции АФК. Это ведёт к окислительному разложению митохондриальных компонентов за счёт накопления супероксидного анион-радикала и к апоптозу [Drane P., Dravard A. Et all, 2001]
Позже, рядом исследователей было изучено влияние активации p53 на экспрессию ряда генов: MnSOD, каталазы, глутатионпероксидазы. Было установлено, что активация p53 приводит к повышению экспрессии MnSOD и глутатионпероксидазы, а экспрессия каталазы не изменяется. Каталаза не будет дисмутировать то количество перекиси водорода, которое образовалось в результате повышенной экспрессии фермента MnSOD. Перекись водорода будет накапливаться в клетках и приводить к апоптозу [Hussain S.P. et all, 2004].
Для исследования динамики экспрессии мРНК гена sod2 выделяли РНК из клеточных культур с p53 (+/+) геномом и с p53 (–/–) при действии ActD.А затем проводили ОТ-ПЦР анализ sod2 и gapdh с последующим электрофорезом полученной ДНК. Полученные результаты экспрессии мРНК гена sod2 нормировали по мРНК гена gapdh, экспрессия которого не изменяется при действии актиномицина D и других воздействиях.
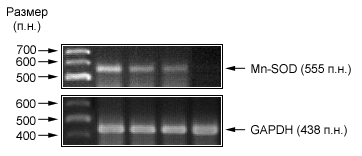
На рис.2 показаны результаты ОТ-ПЦР по генам sod2 и gapdh в клетках MCF-7 до нанесения и через 2 ч,4 ч и 8 ч после нанесения актиномицина D. Из рисунка видно, что при действии ActD экспрессия гена sod2 в клетках MCF7 постепенно снижается и приближается к нулевому значению через 8 часов после нанесения ActD.
На рис.3 показаны результаты ОТ-ПЦР генов MnSOD и gapdh в клетках H1299. Из рисунка видно, что при действии ActD экспрессия гена MnSOD резко снижается в течении первых 2 часов, а затем постепенно восстанавливается, но не достигает контрольного уровня.
На рис. 4 представлены результаты цифровой обработки изображений, показанных на рис. 2 и 3. На рисунке показан график зависимости количества мРНК гена MnSOD в клетках MCF-7 и H1299 от времени нанесения актиномицина D..
Из графика видно, что через 2 часа после нанесения ActD количество мРНК гена MnSOD падает до ~ 45%, а через 4 часа – до ~ 26%. Через 8 часов после нанесения ActD количество МРНК близко к 0%.
В клетках H1299 количество мРНК через 2 часа после нанесения ActD резко упало до ~ 0 %. Через 4 часа после нанесения ActD количество мРНК увеличивается до ~ 10%, а к 8 часам достигает ~ 15%.
Таким образом, в клетках MCF-7 экспрессия мРНК гена MnSOD снижается постепенно. В отличии от резкого падения экспрессии мРНК гена MnSOD в клетках H1299. Вероятно, это объясняется тем, что на ранних этапах действия ActD (в течении 2 и 4 часов) белок p53, присутствующий в клетках MCF-7, в низких концентрациях в течении этого времени (рис.1) препятствует резкому падению экспрессии гена MnSOD, то есть в данном случае белок p53 не столько ингибитором экспрессии гена sod2, сколько фактором,