
Разработка методических основ идентификации избыточных фаз, образующихся в сталях в процессе производства и эксплуатации ответственных изделий машиностроения
| Вид материала | Автореферат |
| Общие выводы |
- Разработка методики снижения вреда окружающей среде при обращении с отходами эксплуатации, 359.08kb.
- Реферат по дисциплине " Технологические процессы микроэлектроники " на тему: Технологические, 1398.5kb.
- «Факторы, формирующие качество швейных изделий. Классификация швейных изделий» Йошкар-Ола, 212.16kb.
- «Технология машиностроения» Специализация «Дизелестроение», 37.35kb.
- Разработка рядового керамического кирпича на базе техногенного отощителя с учетом представлений, 7.06kb.
- Руководство по эксплуатации альбом приложений, 259.28kb.
- Планирование выпуска продукции в условиях рынка. Разработка ассортиментной политики, 204.69kb.
- Особенности алгоритмов идентификации образов человеком и компьютером Владимир И. Андреев, 93.61kb.
- Методические рекомендации по применению примерной номенклатуры дел 4 Список использованных, 433.71kb.
- 6М073000 -производства строительных материалов, изделий и конструкции, 145.77kb.
I110 = I o 110 - I103 - I022 - 0,5I210; I111 = I o 111 - 0,5I210
(пик (210) находится посередине между пиками мартенсита и аустенита и дает одинаковый вклад в их интенсивность). После этого по формулам 1 и 2 определяли количество остаточного аустенита. I и I измеряли по дифрактограммам; Vц определяли по результатам взвешивания карбидных осадков; К=0,75. Результаты приведены в таблице 2.
Как видно из таблицы 4 учет наложения дифракционных пиков цементита на пики матрицы лишь незначительно влияет на конечный результат. Поправка на объемную долю цементита существенна, и ее необходимо учитывать при измерении количества остаточного аустенита в подшипниковых сталях.
Таблица 4. Количество остаточного аустенита (ОА) в образцах стали ШХ15СГ в объемных %.
| № п/п | Образец | ОА (по формуле 1) | ОА с учетом наложения пиков от карбидов | ОА (по формуле 2) |
| 1 | кольцо | 16,0 | 15,7 | 14,6 |
| 2 | кольцо | 16,7 | 16,4 | 15,2 |
| 3 | ролик | 14,9 | 14,6 | 13,3 |
| 4 | ролик | 14,6 | 14,3 | 13,0 |
Определение остаточного аустенита в стали 10Х12Н3М2ФА проводили по методике, изложенной выше применительно к стали ШХ15СГ. Микроструктура металла образца стали 10Х12Н3М2ФА – отпущенный мартенсит. В зависимости от режима отпуска изменялось количество и соотношение вторичных фаз, обнаруженных в образцах методами рентгеноструктурного анализа. Основными фазами были карбид M23C6 и нитрид VN0,35. Общее количество вторичных фаз определяли весовым методом после электролитического выделения анодных осадков, оно составляло 0,5 – 1,0% масс.
Содержание остаточного аустенита менялось от 3% об. в закаленном образце до 0,5% об. в отпущенном. На рисунке 4 приведена дифрактограмма образца с наименьшим содержанием остаточного аустенита – 0,5%. Для сравнения снизу приведены положения дифракционных линий M23C6 и VN0,35 (рассчитаны по дифрактограмме, полученной с электролитически выделенных осадков).
Как видно из рисунка 4, имеется наложение пиков вторичных фаз на пик мартенсита, что может привести к появлению систематической ошибки при определении количества -Fe. Для оценки влияния этого фактора был выбран тот же самый образец с минимальным содержанием остаточного аустенита и максимальным содержанием вторичных фаз. На дифрактограмме (рис. 4) хорошо заметен пик от вторичных фаз. Интегральная интенсивность суммарного пика вторичных фаз – (422) M23C6 плюс частичное наложение (111) VN0,35 – составляет 0,4% от интегральной интенсивности пика (110) -Fe. Интенсивность пика (422) M23C6 равна интенсивности пика (511) M23C6 (соотношение интенсивностей определено по дифрактограмме, полученной с электролитически выделенных осадков), который перекрывается с пиком (110) -Fe. Учитывая все это, можно заключить, что даже в самом неблагоприятном случае максимального содержания вторичных фаз систематической ошибкой, обусловленной наложением дифракционных линий, можно пренебречь. Поправку на объемную долю карбидов и нитридов можно не вносить, поскольку она приведет к появлению коэффициента незначительно отличающегося от единицы ( 0,99).
Методами магнитной ферритометрии и оптической металлографии обнаружить присутствие остаточного аустенита не удалось ни в закаленных, ни в отпущенных образцах стали 10Х12Н3М2ФА. Использование просвечивающей электронной микроскопии позволило выявить отдельные частицы -Fe даже в образцах после отпуска. На рисунке 5 приведено изображение одной из таких частиц. Учитывая результаты, полученные при помощи электронной микроскопии можно понять, почему оптическая металлография не обнаружила аустенит: в мелкодисперсной структуре отпущенного мартенсита достаточно сложно идентифицировать частицы размером порядка 1 мкм.
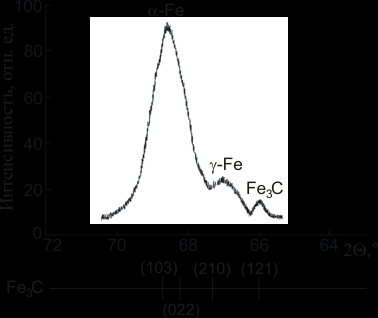
Рис. 3 Рентгенограмма с пиками мартенсита, аустенита и цементита в стали ШХ15СГ (CrK).
Для подтверждения результатов определения количества остаточного аустенита, полученного методом РСФА, были проведены аналогичные измерения с использованием ПЭМ. При увеличении ×10000 было просмотрено 280 полей, что соответствует площади поверхности образца примерно 150 мм2. Количество остаточного аустенита измеряли по площади, занимаемой частицами этой фазы. В результате количество ОА составило 0,7 %, что достаточно хорошо совпадает с содержанием этой фазы, полученным методом РСФА – 0,5%.
Следует отметить, что точность определения количества ОА на пределе чувствительности рентгеноструктурного метода невелика. В условиях эксперимента данной работы при содержании остаточного аустенита 0,5% относительная погрешность составляла 50%. Замена CuK (наиболее распространенное излучение в заводской практике) на излучение хрома может немного повысить разрешение линий мартенсита и аустенита, но существенного влияния на чувствительность и точность измерений не окажет.
Количество остаточного аустенита в металле шва сварного соединения сталей феррито-мартенситного класса является важным технологическим параметром, влияющим на эксплуатационные свойства продукции машиностроительного комплекса. Определение его содержания в сварных соединениях на настоящий момент представляет собой серьезную и не до конца решенную задачу.
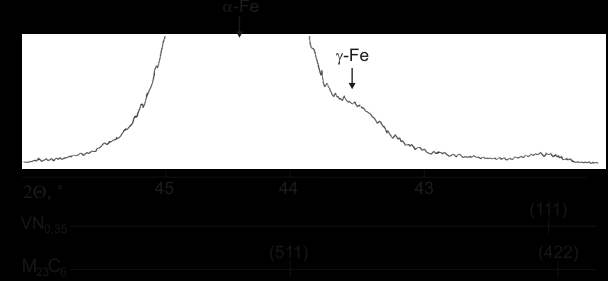
Рис.4 Дифрактограмма образца стали 10Х12Н3М2ФА с минимальным
содержанием остаточного аустенита.

Рис.5 Изображение частицы остаточного аустенита в стали
10Х12Н3М2ФА (ПЭМ, 10000).
Электроды ЦЛ-51 разрабатывали в ЦНИИТМАШ для сварки кавитационно-стойких материалов для гидравлических турбин, и после доработки технологии сварки они нашли применение в атомном энергомашиностроении. Ниже приведены экспериментальные результаты по исследованию сварного соединения двух секторов труб из стали 06Х12Н3Д, изготовленного для моделирования на нем возможных отклонений от штатной термообработки.
Во-первых, это отклонение от штатного режима по температуре второго отпуска – завышение в пределах 30 - 50С. Во-вторых, при изготовлении сварных соединений для зачистки часто используется пневматический инструмент, что приводит к увеличению скорости охлаждения металла шва и, в свою очередь, к изменениям его структуры (фазового состава).
Сварное соединение было разрезано на четыре примерно равные части – плоскости реза были перпендикулярны оси шва. Один образец был термообработан по штатной технологии (режим 1), два с превышением температуры второго отпуска на 30 и 50С (режим 2 и 3 соответственно). Еще один образец был термообработан с повышенной скоростью охлаждения металла шва (охлаждение на воздухе с обдувом после второго отпуска) – режим 4.
Микроструктура металла шва во всех образцах – отпущенный мартенсит и незначительное количество мелкодисперсной карбидной фазы - М23С6. Существенных различий между образцами, подвергнутыми различным термообработкам не выявлено.
Определение содержания остаточного аустенита проводили методами рентгеноструктурного анализа по пикам (111) аустенита и (110) мартенсита. Эти пики были выбраны исходя из соображений изложенных выше. В графическом виде результаты по содержанию ОА приведены на рисунке 6.
Как видно, наибольшее количество остаточного аустенита было обнаружено в образце, термообработанном по штатному режиму (1,9%). В образце, термообработанном по режиму 3 содержание аустенита не превышало чувствительности метода – 0,5%. Общее количество вторичных фаз (М23С6) определяли весовым методом после электролитического выделения анодных осадков, оно составляло 0,5 – 1,0% масс.
Как и в случае со сталью 10Х12Н3М2ФА имеется наложение пиков M23C6 на пик мартенсита, что может привести к появлению систематической ошибки при определении количества -Fe. Интегральная интенсивность пика - (422) M23C6 настолько мала, что не фиксируется на дифрактограмме. Интенсивность пика (422) M23C6 равна интенсивности пика (511) M23C6 (соотношение интенсивностей определено по дифрактограмме, полученной с электролитически выделенных осадков), который перекрывается с пиком (110) -Fe. Учитывая все это, можно заключить, что систематической ошибкой, обусловленной наложением дифракционных линий, можно пренебречь. Поправку на объемную долю карбидов можно не вносить.
Таким образом, метод рентгеноструктурного анализа является единственным методом позволяющим определять небольшие количества (1%) остаточного аустенита в подшипниковых и мартенситных сталях. Это подтверждается хорошей корреляцией полученных результатов по количеству ОА с результатами измерений методом ПЭМ, а так же твердости и ударной вязкости. Другие методы либо не обнаруживают его (оптическая металлография, магнитная ферритометрия), либо могут обнаружить, но из-за большой трудоемкости не могут служить количественным методом (ПЭМ).
Соотношение фаз, образующих матрицу аустенитной стали, оказывает существенное влияние на свойства металлопродукции – физические, механические, коррозионные. Соответственно и идентификация этих фаз является важной задачей, решение которой способствует улучшению качества и продлению сроков эксплуатации изделий машиностроения.
В сталях аустенитного класса возможно образование двух разновидностей -фазы: -феррит и мартенсит (деформационный). Фаза с кристаллической структурой типа оцк может быть достоверно идентифицирована несколькими методами: с помощью оптической металлографии, рентгеноструктурного анализа, магнитной ферритометрии, электронной микроскопии. Однако разделение -феррита и мартенсита при их одновременном присутствии является задачей, пока не имеющей общего решения.
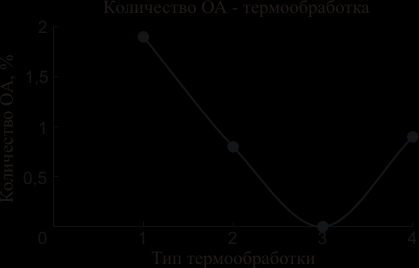
Рис. 6 Содержание ОА металла шва в зависимости от типа термообработки образцов.
Ниже приведены результаты исследования образования дефектов в трубах из сталей 09Х18Н9 и 08Х18Н10Т, которые широко используются в теплообменном энергетическом оборудовании. В ряде поставок от различных производителей на наружной поверхности труб при входном визуальном контроле были обнаружены пятна темно-серого цвета, а в металле были выявлены индикации при токовихревом контроле (ТВК).
В ходе выяснения причин появления дефектов было установлено, что они связаны с образованием ферромагнитной фазы со структурой -Fe. Подобная фаза может быть обусловлена образованием в аустенитной матрице -феррита или деформационного мартенсита. Предложенный в данной работе метод идентификации двух указанных фаз основан на различии механизмов их образования. Во-первых, бездиффузионное мартенситное превращение не приводит к изменению химсостава твердого раствора, т.е. в случае деформационного мартенсита его состав будет идентичен составу аустенита, а состав -феррита будет отличаться большим содержанием ферритообразующих элементов. Во-вторых, деформационный мартенсит образуется на поверхности труб, что обусловлено особенностями технологических операций. Эти две особенности и были положены в основу идентификации образующихся в аустенитных трубах фаз.
Для выяснения пространственного расположения фаз, присутствующих в матрице металла образцов, был использован асимметричный вариант рентгеновской съемки. При этом в некоторых образцах соотношение интенсивностей дифракционных пиков - и -железа изменилось в сторону увеличения интенсивности -Fe. Это свидетельствует о том, что -фаза располагается на поверхности образца, а аустенит располагается под ней. В ряде образцов асимметричная съемка не привела к изменению соотношения интенсивностей. Это свидетельствует о том, что - и -железо равномерно распределены в облучаемом объеме приповерхностного слоя.
Для выявления различия в химическом составе -феррита и деформационного мартенсита был проведен рентгеноспектральный микроанализ участков труб, с которых была получена рентгеновская дифракция, соответствующая оцк решетке -Fe.
Анализ химического состава проводился с поверхности трубы, усреднялись значения на площади 100100 мкм. Полученные результаты приведены в таблице 5.
Таблица 5. Химический состав металла поверхности трубы
| | Si | Mn | Cr | Ni | Ti |
| -фаза -фаза | 0,7 | 0,8 | 18,6 | 9,9 | 0,7 |
| 0,7 | 0,8 | 18,6 | 10,0 | 0,7 |
Как видно их таблицы, химический состав чисто аустенитных участков идентичен составу участков с -Fe. Это свидетельствует о том, что на поверхности ряда образцов трубы образовался деформационный мартенсит. Идентификация фаз, образовавшихся в аустенитных теплообменных трубах, позволила выяснить причины появления дефектов и дать конкретные рекомендации по корректировке технологии изготовления труб.
Использованный комплексный подход (рентгеноструктурный анализ, рентгеноспектральный анализ, ферритометрия, оптическая металлография) позволяет уверенно проводить идентификацию присутствующих в аустените -фаз железа.
В предыдущих разделах были рассмотрены варианты определения фазового состава сталей в том случае, когда одна из компонент является доминирующей. Это определение остаточного аустенита в мартенситных сталях и определение -феррита или деформационного мартенсита в аустенитных сталях. Однако достаточно распространенным представляется случай, когда количества и фаз являются величинами близкими.
Ниже приводятся результаты использования разработанных методов рентгеноструктурного анализа применительно к определению фазового состава дуплексной стали SAF 2205 (02Х22Н5АМ3). Исследования проводили на образцах из ленты 0,788 мм, которая использовалась при изготовления труб для теплообменного оборудования.
В случае ленты кристаллическая текстура соответствует текстуре прокатки. Кроме того, в двухфазной стали каждая из фаз (феррит и аустенит) будет обладать своей собственной текстурой. Возможные текстуры прокатки приведены в таблице 6.
Таблица 6. Текстуры прокатки для аустенита и феррита.
| № п/п | Фаза | Текстура | |
| основная | дополнительная | ||
| 1 | аустенит | {110} 100 | {113} 112 |
| 2 | феррит | {112} 110 {100} 110 | {111} 110 |
Как видно из приведенных данных подобрать дифракционные отражения (кристаллографические плоскости), которые можно использовать для количественного определения соотношения /, при такой сложной текстуре невозможно. Однако избежать влияния кристаллической текстуры в данном случае можно, уменьшив ее интенсивность до приемлемого значения. Достичь этого возможно путем термообработки металла при условии, что она не приведет к увеличению количества -феррита.
В данном разделе решена задача поиска такой термообработки с использованием РСФА, чувствительного к кристаллической текстуре и ОМ, не чувствительной к кристаллической текстуре. Степень интенсивности текстуры определяли в соответствии с критерием ASTM E975. Определение количества феррита рентгеноструктурным методом проводили по методике изложенной выше.
Образцы нагревали до температур, указанных в таблице 7, а затем охлаждали со скоростью 15С/сек для того, чтобы зафиксировать полученное состояние металла.
Таблица 7 Температуры нагрева образцов дуплексной стали
| № образца | 1-1 | 1-2 | 1-3 | 1-4 | 1-5 | 1-6 | 1-7 |
| Температура нагрева,С | 680 | 810 | 960 | 1020 | 1080 | 1300 | 1400 |
В качестве эталонных использовали дифракционные пики -железа (110) и (200), а также пики -железа (111) и (200). Изменение текстуры приводит к изменению соотношения интенсивности линий каждой из фаз, а изменение количества каждой фазы приводит к соответствующему изменению интенсивности линий этой фазы.
Сопоставляя результаты определения соотношения аустенит/феррит, проведенного с помощью двух методов, можно разделить влияние изменения текстуры и количества феррита с аустенитом. Таким образом, сопоставление количества той или иной фазы, полученного методом РСФА и ОМ позволит определить изменения кристаллической текстуры.
Экспериментальные данные представлены в виде графиков зависимости содержания феррита от температуры термообработки на рисунке 7.
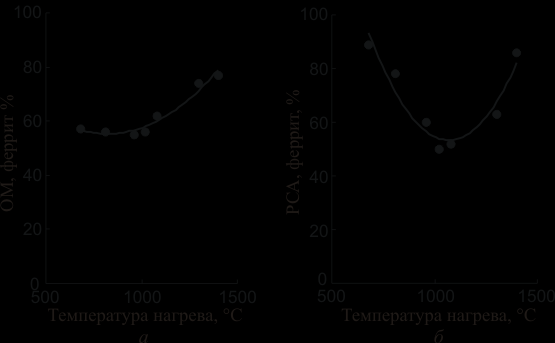
Рис. 7. Зависимость количества феррита от температуры нагрева
а - оптическая металлография; б - рентгеноструктурный анализ.
Как видно из рисунков, зависимости количества феррита от температуры нагрева принципиально различаются для двух методов определения содержания -фазы. В случае использования метода оптической металлографии зависимость содержания феррита от температуры носит монотонный характер. До температуры 1020С количество -фазы практически (в пределах погрешности измерений) не изменяется и только с 1080С начинается существенное увеличение содержания феррита.
При использовании РСФА зависимость содержания феррита от температуры носит явно немонотонный характер. До 1020С происходит уменьшение количества -фазы, а затем начинается рост. Следует обратить внимание на тот факт, что для обоих методов определения содержания феррита существенное увеличение его количества начинается с одной и той же температуры – 1080С.
Изложенные выше экспериментальные данные можно объяснить следующим образом. При использовании метода РСФА существенное искажающее влияние на результаты определения феррита может оказать кристаллическая текстура. В случае применения метода ОМ этого не происходит, т.к. количество -фазы определяется по суммарной площади зерен феррита на окрашенном металлографическом шлифе. Таким образом, до тех пор, пока температура не достигнет значения достаточного для появления ферритной фазы, оптическая металлография не будет фиксировать каких-либо изменений в соотношении фаз.
Полученные результаты позволяют сделать заключение о том, что уменьшение содержания ферритной фазы, фиксируемое рентгеноструктурным методом в интервале температур 680-1020С, обусловлено существенными изменениями в кристаллической текстуре приповерхностных слоев металла. Поскольку определение количества -фазы проводилось по дифракционным линиям (110) феррита, то его снижение означает уменьшение интенсивности пика -фазы. Это означает, что интенсивность первоначальной кристаллической текстуры, при которой кристаллографическая плоскость (110) элементарной ячейки феррита была параллельна поверхности металла, существенно ослабевала с ростом температуры. При температуре 1020С, отвечающей минимальному количеству феррита, кристаллическая текстура практически исчезала. Дальнейший рост содержания феррита, фиксируемый и оптической металлографией и рентгеноструктурным методом, обусловлен только фазовыми превращениями и не связан с кристаллической текстурой металла.
Таким образом, методы РСФА применимы по отношению к определению соотношения феррит/аустенит в дуплексных сталях типа SAF 2205(02Х22Н5АМ3) после термообработки при температуре не ниже 1020С. Следует отметить, что эта температура ниже той, при которой происходит термообработка труб из дуплексной стали после процесса сварки – 1050С [129,130]. Этот факт означает, что контроль соотношения феррит/аустенит, как для основного металла труб, так и для сварных соединений можно проводить методами рентгеноструктурного анализа.
Для подтверждения этого было проведено определение соотношения / в металле шва труб 280,7 мм из указанной выше стали методами ОМ и РСФА. Обработка металлографического изображения показала, что содержания феррита и аустенита равны и составляют 50%. Рентгеноструктурный анализ дал практически аналогичные результаты 48% феррита и 52% аустенита.
ОБЩИЕ ВЫВОДЫ
- Разработан метод дифференциального рентгеноструктурного фазового анализа сталей. Он базируется на разделении избыточных фаз различными методами (химический, гравиметрический, магнитный и т.д.), что существенно повышает достоверность проводимого анализа. В результате последовательного удаления фаз, содержащихся в электролитически выделенных осадках, достигается повышение чувствительности и разрешающей способности рентгеноструктурного фазового анализа. Чувствительность возрастает после удаления преобладающей фазы, разрешающая способность – при уменьшении числа наложений дифракционных линий.
- Экспериментально при проведении исследований большого числа разнообразных сталей, используемых в машиностроении, подтверждена высокая надежность разработанного метода. Результаты рентгеноструктурного анализа проверялись иными методами исследования фазового состава, в частности оптической металлографией, просвечивающей и растровой электронной микроскопией, рентгеноспектральным микроанализом, карбидным анализом. В каждом случае получено хорошее совпадение результатов РСФА с результатами перечисленных методов.
- На основании проведения экспериментальных исследований, осуществленных для широкого круга сталей, определены оптимальные варианты методов и условия проведения идентификации и анализа фаз. Варианты обусловлены химическим составом сталей, особенностями технологического цикла изготовления и режимами эксплуатации металлопродукции или изделий машиностроения.
- Разработанный метод был применен в экспериментальных исследованиях избыточных фаз, образующихся в сталях в процессе изготовления и эксплуатации ответственных изделий машиностроения, что позволило впервые методами рентгеноструктурного анализа:
- идентифицировать в низколегированных сталях весь комплекс избыточных фаз вплоть до наночастиц спецкарбидов, формирующихся в процессе изготовления и длительной эксплуатации;
- выявить в высокохромистых роторных сталях наночастицы карбида ванадия со сложной гексагональной кристаллической структурой;
- идентифицировать пленочные наночастицы карбида ниобия, образовавшиеся в трубах из аустенитной стали после длительной эксплуатации на тепловых станциях
- Развит метод рентгеноструктурного анализа для проведения количественного определения остаточного аустенита, который позволяет учесть влияние кристаллической текстуры и выделения избыточных фаз на результаты измерений. Показано, что влияния кристаллической текстуры можно избежать, используя определенные пары дифракционных пиков, выбранных с учетом возможных ориентационных соответствий, действующих при фазовых превращениях в сталях. Негативное воздействие избыточных фаз можно исключить, используя анодные осадки, выделенные непосредственно из исследуемых образцов.
- Путем проведения экспериментальных исследований различного типа сталей, подтверждена высокая достоверность предлагаемого метода определения остаточного аустенита. Результаты рентгеноструктурного анализа проверялись методами оптической металлографии, просвечивающей электронной микроскопии, магнитной ферритометрии. Для всех случаев получено хорошее соответствие результатов РСФА и указанных методов.
- Экспериментально исследованы изменения состава матрицы сталей аустенитного и аустенито-ферритного классов изготовленных с использованием различных технологических процессов при изготовлении и воздействия различных факторов в процессе эксплуатации.
- Полученные в результате использования разработанных методов идентификации и контроля фазового состава сталей новые данные были использованы при разработке и оптимизации технологий производства ответственных изделий машиностроения.