
Магазины электрических величин
| Вид материала | Документы |
- Рабочей программы дисциплины Электроэнергетические системы и сети по направлению подготовки, 21.71kb.
- Отчет по лабораторной работе должен содержать: наименование работы и номер, схемы, 365.83kb.
- Экзаменационные вопросы по курсу «Электротехника и электроника», 23.91kb.
- Бизнес-план магазина товаров для детей Содержание, 138.19kb.
- 1. Основные понятия и обозначения электрических величин и элементов электрических цепей., 277.03kb.
- Цифровой вольтметр щ-304, 137.06kb.
- Телемеханики, 26.01kb.
- Отдел метрологического обеспечения измерений электрических величин, 42.58kb.
- Курсовая работа по курсу «основы физических измерений», 226.86kb.
- Теория электрических цепей (часть, 63kb.
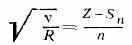
(R — Ридберга постоянная, Sn — постоянная экранирования, учитывающая влияние на отдельный эл-н всех остальных эл-нов атома, n — главное квантовое число (см. Квантовые числа). Установлен экспериментально англ.
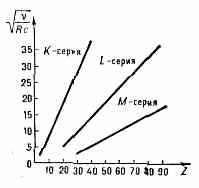
физиком Г. Мозли (Н. Moseley) в 1913. На диаграмме Мозли (рис.) зависимость v от Z представляет собой ряд прямых (К-, L-, М- и т. д. серии, соответствующие n=1, 2, 3, . . .). В каждой серии при переходе от Z к Z+1 значение v увеличивается на одну и ту же величину, благодаря этому элементы можно расположить в ряд в соответствии с возрастанием Z. Исторически М. з. позволил окончательно подтвердить, что Z определяется зарядом ядра, а не ат. массой. Это устранило последние сомнения в правильности размещения элементов в периодической системе элементов.
А. В. Колпаков.
МОЛЕКУЛА (новолат. molecule, уменьшит. от лат. moles — масса), наименьшая ч-ца в-ва, обладающая его осн. хим. св-вами и состоящая из атомов, соединённых между собой химическими связями. Число атомов в М. составляет от двух (Н2, О2, HF, KCl)
430
до сотен и тысяч (нек-рые витамины, гормоны и белки). Атомы инертных газов часто называют одноатомными М., хотя, строго говоря, они не явл. М. Если М. состоит из тысяч и более повторяющихся единиц (одинаковых или близких по строению групп атомов), то её называют макромолекулой. В физике представление о М. возникло в 18 в. и получило широкое признание в 19 в. в связи с развитием термодинамики и теории газов и жидкостей. Во 2-й половине 19 в. с помощью разл. хим. методов были получены мн. важные сведения о строении М. Окончательно существование М. было подтверждено опытами франц. физика Ж. Б. Перрена по изучению броуновского движения (1906).
Атомы в М. связаны между собой в определ. последовательности и определ. образом расположены в пр-ве. Наиб. общие хар-ки М.— мол. масса, состав и структурная ф-ла, указывающая последовательность хим. связей (напр., мол. масса М. воды 18, равная сумме масс входящих в неё атомов в атомных единицах массы, состав Н2О, структурная ф-ла Н—О—Н). Прочность межатомной связи характеризуется энергией хим. связи, к-рая составляет обычно неск. десятков кДж/моль. Атомы в М. непрерывно совершают колебат. движения; при определ. условиях, напр. в газовой фазе, М. могут совершать поступат. и вращат. движения. М., как и атомы, не имеют чётких границ. Размеры М. можно ориентировочно оценить, зная плотность в-ва, мол. м. и число Авогадро. Так, если допустить, что М. Н2O имеет сферич. форму, то диаметр её окажется равным ~3•10-8 см (0,3 нм). Размеры М. растут с увеличением числа атомов в них и лежат в пределах 10-8—10-5 см. М. нельзя увидеть невооружённым глазом или с помощью оптич. микроскопа, однако существование М. доказывают мн. явления (броуновское движение, диффузия, дифракция рентг. лучей, эл-нов, нейтронов и т. д.).
Устойчивость М. в среде зависит от её вз-ствия с др. атомами, а также от темп-ры, давления и др. внеш. факторов. В газообразном состоянии в-во, как правило, состоит из М. (кроме инертных газов, паров металлов). При достаточно высоких темп-pax М. всех газов распадаются на атомы. В конденсированных системах М. могут сохраняться. Вода во всех агрегатных состояниях состоит из М.; из М. построены большинство жидкостей и молекулярные кристаллы. В металлах и др. ат. кристаллах, а также их расплавах М., как правило, не существуют, т. к. в них каждый атом взаимодействует со всеми соседними приблизительно одинаково.
Химическая связь. Возможность образования М. объясняется тем, что внутр. энергия М. как системы атомов ниже суммарной энергии этих атомов в изолиров. состоянии. Соответствующая разность энергии наз. энергией образования М. из атомов (или энергией атомизации), к-рая приближённо равна сумме энергий хим. связей.
Для хим. связи существ. значение имеют лишь эл.-магн. вз-ствия эл-нов и ядер входящих в М. атомов. Наиболее часто встречаются М., в к-рых существуют ковалентные и ионные хим. связи.
К о в а л е н т н а я с в я з ь возникает при обобществлении эл-нов (обычно электронных пар) двумя соседними атомами (т. е. за счёт обмена эл-нами). Хим. связь такого типа осуществляется в М. Н2, O2, СО и др. При сближении атомов ковалентная связь образуется только в том случае, когда спины их внеш. эл-нов антипараллельны. При этом происходит деформация электронных оболочек атомов, их перекрытие по линии, соединяющей ядра. При нек-ром межъядерном расстоянии силы притяжения уравновешиваются силами отталкивания и образуется устойчивая система, внутр. энергия к-рой минимальна.
Ионная связь осуществляется электростатич. вз-ствием атомов при переходе эл-на одного из них к другому, т. е. при образовании положит. и отрицат. иона. Такая связь характерна для М. NaCl, KI и др. Ковалентные и ионные хим. связи явл. предельными; как правило, образуются смешанные хим. связи — частично ковалентные, частично ионные.
Внутренняя энергия и уровни энергии молекул. Внутр. энергия М.— осн. хар-ка, определяющая её состояние и св-ва и зависящая от взаимного расположения составляющих её ч-ц и их движения. М. явл. квант. системой, и её внутр. энергия ξ может принимать лишь определ. значения, т. е. квантуется. Внутр. энергия М. приближённо равна сумме энергий электронных движений ξэ, колебаний ядер ξк и вращения М. как целого ξв, т. е. ξξэ+ξк+ξв, причём ξэ>>ξк>>ξв. Каждая из указанных энергий квантуется в соответствии с законами квантовой механики, и ей соответствует набор дискретных уровней энергии (электронные, колебат. и вращат. уровни энергии).
Состояние М. как квант. системы описывается Шредингера уравнением, к-рое учитывает электростатич. вз-ствия эл-нов с ядрами, эл-нов друг с другом, а также кинетич. энергию эл-нов и ядер. В адиабатическом приближении ур-ние Шредингера для М. распадается на два ур-ния — для эл-нов и для ядер. Решение (обычно приближённое) электронного ур-ния Шредингера — нахождение уровней энергии эл-нов — одна из осн. задач квантовой химии.
М.— электрически нейтральные системы, однако электронная плотность в них распределена неравномерно. Число электронных уровней в М.
значительно больше числа уровней энергии составляющих М. атомов, поскольку каждый атом М. находится в электрич. поле остальных атомов, в результате чего уровни расщепляются на многочисл. подуровни (Штарка эффект).
Электронные уровни М. определяются совокупностью квантовых чисел, характеризующих состояния всех эл-нов М. Уровни, отвечающие значениям квант. числа =0, 1, 2, ... полного орбит. момента М обозначаются соответственно , П, , ... ( представляет собой сумму орбитальных квант. чисел эл-нов; см. Атом). Квант. число S=0, 1, 2, ... определяет полный спиновый момент, внутр. квант. число =r±S— полный момент М. Электронный уровень М. обозначают 2S+1, где слева вверху приводится мультиплетность уровня =2S+1.
Ур-ние Шредингера для ядер содержит информацию о колебаниях М. и вращениях её как целого. Решение этого ур-ния для двухатомной М. приводит к дискретным колебат. уровням, отстоящим один от другого на hv, если колебания ядер считать гармоническими (v — собств. частота осциллятора), и на hv-2(v+1)hva — при ангармонич. колебаниях (v — колебательное квант. число, а — постоянная ангармоничности). Колебания реальных двухатомных М. ангармоничны, и расстояние между колебат. уровнями энергии убывают с ростом v, а макс. колебат. энергия равна энергии диссоциации М.
В многоатомной М. как связанной системе ч-ц колебания отд. атомов не независимы. Сложные колебания такой системы можно разделить на независимые гармонич. колебания, каждое из к-рых характеризуется определ. частотой; их называют н о р м а л ь н ы м и к о л е б а н и я м и.
Колебания многоатомных М. в принципе могут быть изучены теоретически с помощью методов квант. химии, однако на практике обычно пользуются механич. моделью, оперирующей силовыми постоянными разл. структурных элементов М.
Вращат. уровни двухатомной М. определяются выражением ξв= h2J(J+1)/82I, где I — момент инерции М., относительно нек-рой оси вращения, J — вращат. квант. число. Аналогичные ф-лы, выведенные для многоатомных М., позволяют определять их геометрию по наблюдаемым чисто вращат. спектрам. Выражение для ξв резко усложняется, если, помимо вращения М. как целого, имеет место внутр. вращение, приводящее к ротамерам (см. ниже). Однако ф-лы для ξв дают возможность на основании вращат. спектров оценивать барьеры внутр. вращения и др. хар-ки М. Наряду с чисто элект-
431
ронными, колебат. и вращат. уровнями энергии в спектрах проявляются уровни, обусловленные электронно-колебат. и колебательно-вращат. вз-ствиями.
Спектры излучения, поглощения, комбинац. рассеяния света возникают при переходах М. с одного уровня энергии на другой; при этом М. поглощает или излучает энергию, равную разности энергий этих уровней. Соответственно возникают электронные, колебат. и вращат. спектры М. (подробнее см. Молекулярные спектры).
Структура молекулы. Геометрию М. можно описать декартовыми координатами атомов, однако чаще всего её характеризуют набором внутр. параметров — д л и н с в я з е й, в а л е н т н ы х и д в у г р а н н ы х у г л о в. Длиной связи наз. расстояние между ядрами атомов, соединённых между собой хим. связью. Обычно, чем больше длина связи, тем меньше её прочность.
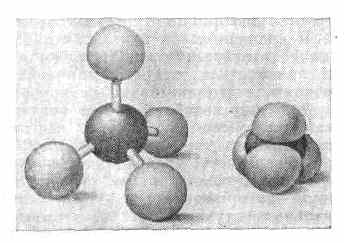
Трёхмерные модели молекул: слева — шаро-игловая модель: атомы изображены белыми шарами, валентные связи — выходящими из них стержнями; справа — объёмная модель Стюарта—Бриглеба: атомы представлены шарами, радиусы к-рых пропорц. ван-дер-ваальсовым радиусам.
Валентным наз. угол между двумя хим. связями, выходящими из одного атома. Торсионные углы — это углы вращения вокруг связей. Так, в М. перекиси водорода, Н—О—О — Н, длины связей О—О и О—Н равны соотв. 0,147 и 0,095 нм, валентный угол Н—О—О равен 95° и торсионный угол (угол вращения вокруг связи О—О, или двугранный угол между плоскостями Н—О—О и О—О—Н) равен 112°.
Каждое электронное состояние характеризуется равновесной геометрией (равновесной конфигурацией), отвечающей мин. энергии. В обычных условиях М. находится в основном электронном состоянии (на ниж. электронном уровне), и термин «равновесная конфигурация» часто относят только к этому состоянию. Так, приведённые выше внутр. геом, параметры М. Н—О — О — Н явл. равновесными, тогда как, напр., плоские формы этой М. (торсионный угол равен 0 или 180°) неравновесны. Зависимость внутр. энергии М. от геом. параметров для многоатомных М. может быть представлена многомерной поверхностью, наз. потенциальной поверхностью. Самый глубокий минимум потенц. энергии М. соответствует её равновесной конфигурации, метастабильным состояниям отвечают менее глубокие минимумы. Определение потенц. поверхности М. или хотя бы выявление нек-рых её особенностей явл. целью разл. эксперим. и теоретич. исследований.
Расположение атомов в М. всегда обладает определённой симметрией (см. Симметрия молекулы). Потенц. поверхность М. также обладает симметрией, что проявляется, напр., в инфракрасных спектрах М. или спектрах комбинационного рассеяния света.
Нек-рые одинаковые по составу М. могут отличаться строением или расположением атомов. Такие формы существования в-ва наз. изомерами (см. Изомерия молекул). Структурные изомеры имеют разную последовательность хим. связей, и их М. изображаются разными структурными ф-лами (напр., нормальный бутан Н3С—СН2—СН2—СН3 и изобутан
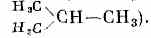
Структурные изомеры — это разные М., а соответствующие соединения обладают разными физ. и хим. св-вами. Так, темп-ра кипения нормального бутана равна +0,6 °С, а изобутана -11,7 °С.
Поворотные изомеры (р о т а м е р ы, к о н ф о р м е р ы) возникают при вращении атомов или ат. групп вокруг хим. связей и отвечают разным минимумам потенц. поверхности М. Они представляют собой разл. состояния одной и той же М. Энергетич. барьеры, разделяющие поворотные изомеры, не превышают 100 кДж/моль, а время жизни этих изомеров обычно ~10-10—10-13 с. При более высоких энергетич. барьерах (напр., при геом. изомерии М.) время жизни изомеров возрастает и появляется возможность их разделения.
М. оптических изомеров энантиоморфны — зеркально симметричные одна по отношению к другой. Такие изомеры вращают плоскость поляризации света в противоположные стороны; остальные же физ. св-ва у них совершенно одинаковы (см. Оптически активные вещества).
Свойства молекул. Исследование молекул. Хим. и большинство физ. св-в М. определяются их внеш. эл-нами. Так, внеш. эл-ны определяют оптич. спектры М. В спектрах М. проявляются мн. особенности их строения (симметрия, изомерия, природа хим. связи и т. д.). Индивидуальность спектров соединений, характеристичность частот колебаний определ. групп атомов в М. позволяют производить качеств. и количеств. спектральный анализ в-ва. Электронные уровни энергии изучают методами ультрафиолетовой спектроскопии, фотоэлектронной спектроскопии, рентгеноэлектронной спектроскопии. Колебат. уровни энергии проявляются в ИК спектрах и спектрах комбинац. рассеяния света. Частоты вращат. линий лежат в радиодиапазоне (см. Микроволновая спектроскопия), а также в дальней ИК области спектра.
Во внеш. электрич. поле М. поляризуется — приобретает индуциров. дипольный момент (см. Поляризуемость атомов, ионов, молекул). Поляризация в-в из полярных М. (т. е. М., обладающих пост. дипольными моментами) во внеш. электрич. поле обусловлена как их ориентацией вдоль поля, так и возникновением индуциров. дипольных моментов за счёт электронной поляризуемости. Измерение диэлектрич. проницаемости и поляризации в-ва даёт возможность приблизительно оценивать поляризуемость и величину пост. дипольных моментов отд. М., что позволяет делать выводы о её строении — симметрии, распределении электронной плотности, присутствии тех или иных групп атомов и их расположении и т. д.
Магн. св-ва М. дают важные сведения о строении электронной оболочки. Большинство М. диамагнитны, т. е. не имеют пост. магн. момента. Поведение таких М. в магн. поле определяется их отрицат. магнитной восприимчивостью. Парамагн. М., обладающие пост. магн. моментом, во внеш. магн. поле стремятся ориентироваться в направлении поля. Пост. магн. моментом (связанным со спином эл-нов, а также с их орбит. движением) могут обладать как электронная оболочка, так и ат. ядра. Парамагнитные (обладающие неспаренным эл-ном) М. исследуют с помощью электронного парамагнитного резонанса. В спектрах ядерного магнитного резонанса проявляются вз-ствия спиновых моментов ат. ядер, зависящие от электронной структуры М. и окружения каждого атома. На основании спектров ЯМР судят о направлении хим. связей, различных проявлениях изомерии М., взаимном расположении атомов в М., о динамике атомов в М. и т. д.
Важный метод изучения М.— массспектроскопия. Масс-спектрометрич. измерения основаны на расщеплении М. на электрически заряж. фрагменты (радикалы) и определении масс этих фрагментов. Геометрию М. в кристаллах определяют с помощью дифракции рентг. лучей (см. Рентгеновский структурный анализ) и нейтронов (см. Нейтронография). В газовой фазе и парах геометрию М. исследуют с помощью дифракции эл-нов (см. Электронография) и микроволновых спектров. Эти исследования дают точность в определении координат атомов (ядер) порядка 0,001 нм; отсюда точность в определении длин связей ~0,001 нм и в определении
432
валентных и двугранных углов — 1—2°. Помимо дифракц. и спектроскопич. методов, существует ещё ряд методов исследования структурных, динамич. и термодинамич. хар-к М. Так, термодинамич. методы (в частности, калориметрия) позволяют определять разность энтальпий разл. изомеров, поглощение УЗ используется для установления равновесного содержания изомеров в жидкостях и р-рах и т. д.
• Волькенштейн М. В., Строение и физические свойства молекул, М.—Л., 1955: Картмелл Э., Фоулз Г. В. А., Валентность и строение молекул, пер. с англ., М., 1979; Ельяшевич М. А., Атомная и молекулярная спектроскопия, М., 1962; Ландау Л. Д., Л и ф ш и ц Е. М., Квантовая механика. Нерелятивистская теория, 3 изд., М., 1974 (Теоретическая физика, т. 3).
В. Г. Дашевский.
МОЛЕКУЛЯРНАЯ АКУСТИКА, раздел физ. акустики, в к-ром св-ва в-ва и кинетика мол. процессов исследуются акустич. методами. Осн. методами М. а. явл. измерение скорости звука и поглощения звука и зависимостей этих величин от частоты звук. волны, темп-ры, давления и др. Методами М. а. можно исследовать газы, жидкости, полимеры, тв. тела, плазму.
По скорости звука можно определить сжимаемость, отношение теплоёмкостей cp/cV, модули упругости тв. тела и др., а по поглощению звука — значение сдвиговой вязкости и объёмной вязкости, время релаксации р и др. В газах, измеряя скорость звука и её зависимость от темп-ры, определяют параметры, характеризующие вз-ствие молекул газа при столкновении. В жидкостях, вычисляя скорость звука на основании той или иной модели жидкости и сравнивая результаты расчёта с опытными данными, в ряде случаев можно оценить правдоподобность используемой модели и определить энергию вз-ствия молекул. На скорость звука влияют особенности мол. структуры, силы межмолекулярного взаимодействия и плотность упаковки молекул. Так, напр., увеличение плотности упаковки молекул, появление водородных связей, полимеризация приводят к увеличению скорости звука, а введение в молекулу тяжёлых атомов — к её уменьшению.
При наличии релаксац. процессов (см. Релаксация акустическая) энергия поступат. движения молекул в звук. волне перераспределяется на внутр. степени свободы. При этом появляется дисперсия звука, а зависимость произведения коэфф. поглощения на длину волны от частоты звука имеет максимум на нек-рой частоте р=1/р, наз. частотой релаксации. Величины дисперсии звука и коэфф. поглощения зависят от того, какие именно степени свободы возбуждаются под действием звук. волны, а р связана со скоростью обмена энергией между разл. степенями свободы. Т. о., измеряя скорость звука
и коэфф. поглощения в зависимости от частоты звука и определяя р, можно судить о характере мол. процессов и о том, какой из них вносит осн. вклад в релаксацию. Эти методы позволяют исследовать возбуждение колебат. и вращат. степеней свободы молекул в газах и жидкостях, столкновение молекул в смесях разл. газов, установление равновесия при хим. реакциях, перестройку мол. структуры в жидкостях, процессы сдвиговой релаксации в очень вязких жидкостях и полимерах, разл. процессы вз-ствия звука с эл-нами проводимости, фононами и др. элем. возбуждениями в тв. телах.
Область релаксации для жидкостей лежит, как правило, в диапазоне более высоких частот, чем для газов. В очень вязких жидкостях, полимерах и нек-рых др. в-вах в поглощение и дисперсию может давать вклад целый набор релаксац. процессов с широким спектром времён релаксации. Изучение температурных зависимостей скорости и поглощения звука позволяет разделить вклад разл. релаксац. процессов.
В М. а. для исследований обычно применяется УЗ: в газах — в диапазоне частот 104—105 Гц, а в жидкостях и тв. телах — в диапазоне 105— 1010 Гц.
Методы М. а. могут быть также использованы для исследования таких в-в, в к-рых вз-ствие звука с элементарными возбуждениями не ограничивается простейшими релаксац. процессами. Так, напр., методы М. а. используются для исследования кинетики мол. процессов в критич. области. Исследования поглощения УЗ в металлах и ПП при разных темп-pax, магн. полях и др. внеш. воздействиях позволяют получить информацию о поведении эл-нов и об особенностях электрон-фононного вз-ствия. Измерение внутр. трения в диэлектриках, напр. в кварце, в зависимости от темп-ры и при разных условиях предварит. обработки позволяет судить о наличии тех или иных примесей и дефектов.
• Михайлов И. Г., Соловьев В. А., Сырников Ю. П., Основы молекулярной акустики, М., 1964; Физическая акустика, под ред. У. Мэзона, пер. с англ., т. 2, ч. А, М., 1968; т. 4, ч. А—Б, М., 1969—70; т. 5, 7, М., 1973—74.
А. Л. Полякова.
МОЛЕКУЛЯРНАЯ МАССА, значение массы молекулы, выраженное в атомных единицах массы. Практически М. м. равна сумме масс входящих в неё атомов (см. Атомная масса).
МОЛЕКУЛЯРНАЯ ФИЗИКА, раздел физики, в к-ром изучаются физ. св-ва тел в разл. агрегатных состояниях на основе рассмотрения их микроскопич. (молекулярного) строения. Задачи М. ф. решаются методами физ. статистики, термодинамики и физ. кинетики, они связаны с изучением движения и вз-ствия ч-ц (атомов, молекул, ионов), составляющих физ. тела.
Первым сформировавшимся разделом М. ф. была кинетическая теория газов. В процессе её развития работами англ. физика Дж. Максвелла (1858—60), австр. физика Л. Больцмана (1868) и амер. физика Дж. У. Гиббса (1871 —1902) была создана классич. статистич. физика.
Количеств. представления о вз-ствии молекул (мол. силах) начали развиваться в теории капиллярных явлений. Классич. работы в этой области франц. учёных А. Клеро (1743), П. Лапласа (1806), англ. учёного Т. Юнга (1805), франц. учёного С. Пуассона, нем. учёного К. Гаусса (1830 — 1831), Гиббса (1874—78), И. С. Громеки (1879, 1886) и др. положили начало теории поверхностных явлений. Межмол. вз-ствия были учтены голл. физиком Я. Д. Ван-дер-Ваальсом при объяснении физ. св-в реальных газов и жидкостей.
В нач. 20 в. М. ф. вступает в новый этап развития. В работах франц. физика Ж. Б. Перрена и швед. учёного Т. Сведберга (1906), польск. физика М. Смолуховского и А.. Эйнштейна (1904—06), посвящённых броуновскому движению микрочастиц, были получены доказательства реальности существования молекул. Методами рентгеновского структурного анализа (а впоследствии — методами электронографии и нейтронографии) были изучены структура тв. тел и жидкостей и её изменения при фазовых переходах и изменении темп-ры, давления и др. хар-к. Учение о межатомных взаимодействиях на основе представлений квантовой механики получило развитие в работах нем. физиков М. Борна, Ф. Лондона и В. Гейтлера, а также П. Дебая (Германия). Теория переходов из одного агрегатного состояния в другое, намеченная Ван-дер-Ваальсом и англ. физиком У. Томсоном и развитая в работах Гиббса, Л. Д. Ландау (1937) и нем. физико-химика М. Фольмера (30-е гг.) и их последователей, превратилась в современную теорию образования фазы — важный самостоят. раздел М. ф. Объединение статистич. методов с совр. представлениями о структуре в-ва в работах Я. И. Френкеля, англ. физико-химика Г. Эйринга (1935—36), англ. учёного Дж. Бернала и др. привело к М. ф. жидких и тв. тел.
Круг вопросов, охватываемых М. ф., очень широк. В ней рассматриваются : строение в-в и его изменение под влиянием внеш. факторов (давления, темп-ры, электрич. и магн. полей), явления переноса (диффузия, теплопроводность, внутр. трение), фазовое равновесие и процессы фазовых переходов (кристаллизация и плавление, испарение и конденсация и др.), критич. состояние в-ва, поверхностные явления на границах раздела разл. фаз.
433
Интенсивное развитие М. Ф. привело к выделению из неё мн. самостоят. разделов (статистич. физика, физ. кинетика, физика тв. тела, физ. химия, мол. биология). На основе общих теоретич. представлений М. ф. получили развитие физика металлов, физика полимеров, физика плазмы, кристаллофизика, физико-химия дисперсных систем и поверхностных явлений, теория массо- и теплопереноса, физико-хим. механика. При всём различии объектов и методов исследования здесь сохраняется. однако, гл. идея М. ф.— описание макроскопич. св-в в-ва на основе микроскопической (молекулярной) картины его строения.
• Кикоин И. К., Кикоин А. К., Молекулярная физика, 2 изд., М., 1976; Гиршфельдер Дж., К е р т и с с Ч., Берд Р., Молекулярная теория газов и жидкостей, пер. с англ., М., 1961; Френкель Я. И., Кинетическая теория жидкостей, Л., 1975; Лихтман В. И., Щ у к и н Е. Д., Ребиндер II. А., Физико-химическая механика металлов, М., 1962.
П. А. Ребиндер, Б. В. Дерягин, Н. В. Чураев.