Качан Александр Вячеславович Руководители профессор, доктор биол наук Евтушенков А. Н. ст преподаватель Апанасевич Т. А. Минск, 2010 Оглавление Оглавление 3 реферат
| Вид материала | Реферат |
- Апанасевич Татьяна Антоновна Минск 2010 г. Оглавление Оглавление 3 Список условных, 450.52kb.
- Сидорская Ирина Владимировна старший преподаватель Кожич Павел Павлович Минск 2010, 900.44kb.
- Коледа Виктор Антонович, ст преподаватель Пол Кожич Минск 2010 г. Оглавление Оглавление, 340.48kb.
- Груша Александр Иванович, старший преподаватель Воробьев Михаил Алексеевич Минск 2010, 248.05kb.
- Краснопрошин Виктор Владимирович ст преподаватель Кожич Павел Павлович Минск 2008, 166.87kb.
- Кожич Павел Павлович, профессор Воробьев Василий Петрович Минск 2010 г. Оглавление, 247.09kb.
- Кожич Павел Павлович Минск 2010 г Оглавление Оглавление 2 Применение информационных, 302.47kb.
- Писарев Валерий Михайлович, Ст преп. Воробьев М. А. Минск 2010 оглавление оглавление, 415.9kb.
- Кожич Павел Павлович, доцент Запрудский Сергей Николаевич Минск 2010 г. Оглавление, 202.51kb.
- Бокун Наталья Чеславовна Ассистент: Шешко Сергей Михайлович Минск 2008 оглавление оглавление, 270.07kb.
Белорусский государственный университет
Выпускная работа по
“Основам информационных технологий”
Аспирант
биологического факультета
кафедры молекулярной биологии
Качан Александр Вячеславович
Руководители
профессор, доктор биол. наук Евтушенков А. Н.
ст. преподаватель Апанасевич Т. А.
Минск, 2010
Оглавление
Оглавление 3
Реферат по информационным технологиям на тему: «Использование информационных технологий в анализе структуры и функций белков»
ВВедение 5
Глава 1. Обзор литературы 8
1.1 Изучение первичной структуры 8
1.2 Моделирование трёхмерной структуры белков 8
1.3 Функциональная протеомика 10
Глава 2. Методика исследования белковых последовательностей 12
2.1 Общие базы данных 12
2.2 Специализированные базы данных 13
2.3 Сравнение последовательностей 13
2.4 Моделирование вторичной структуры 15
Глава 3. Компьютерный анализ аминокислотной последовательности гена амилазы штамма Bacillus sp. 406 16
3.1 Обнаружение гомологичных белков и классификация 16
3.2 Обнаружение сигнальных последовательностей 19
3.3 Анализ физико-химических параметров белка 19
3.4 Моделирование вторичной структуры 20
3.5 Эволюционный анализ 22
Заключение 24
Предметный указатель 26
Список Иллюстраций 27
Список использованной литературы 28
Интернет-ресурсы в предметной области 29
Действующий личный сайт в Интернет 32
Граф научных интересов 33
Презентация 34
Список литературы по выпускной работе 35
Приложение 36
Приложение 37
Реферат по информационным технологиям на тему: «Использование информационных технологий в анализе структуры и функций белков»
ВВедение
Во второй половине XX в. бурно развивались аналитические методы биохимии, молекулярной биологии и вычислительной техники. Выдающиеся успехи, достигнутые в этих областях, привели к возможности расшифровки огромных последовательностей оснований нуклеиновых кислот и к записи полного генома живого организма. Впервые полный геном был расшифрован в 1980 г. у бактериофага φХ-174, затем у первой бактерии – Haemophilus influenzae. На рубеже XX-XXI веков была закончена работа по расшифровке полного генома человека – последовательности из примерно 3 млрд оснований. Всего же уже расшифрованы геномы нескольких десятков видов живых организмов. Именно в этот период возникли две новые биологические науки: в 1987 г. впервые в научной печати было использовано слово «геномика», а в 1993 г. – «биоинформатика». Позже к этим областям знаний также добавляется протеомика – наука, занимающаяся изучением совокупности белков и их взаимодействий в живых организмах [4].
Под биоинформатикой обычно понимают изучение биологической информации с помощью математических, статистических и компьютерных методов. В настоящее время это почти исключительно задачи молекулярной биологии. Причина этого в том, что за последние 20–25 лет накоплен огромный экспериментальный материал именно о строении и функционировании биологических молекул (белков и нуклеиновых кислот) самых разных организмов. Этот материал требует развитых компьютерных методов для своего анализа. Поэтому биоинформатика в первую очередь понимается как синоним вычислительной молекулярной биологии. Более того, без компьютерной обработки, моделирования и анализа данных решение большинства задач геномики и протеомики просто невозможно [3].
Исследование белков с помощью средств биоинформатики имеет особую важность по нескольким причинам. Во-первых, белки способны выполнять в клетке самые разнообразные биологические функции. При этом общий состав клеточных белков постоянно меняется в зависимости от фазы клеточного цикла, тканевой принадлежности и стадии дифференцировки, внешних воздействий. Кроме того, помимо набора, меняется еще и количество белков: от нескольких молекул до нескольких тысяч на клетку. Основной задачей протеомики является предсказание функциональной роли отдельных белков путем экспериментального сопоставления их качественного и количественного состава в клетке на разных стадиях и в разных состояниях ее развития.
Во-вторых, в задачу протеомики входит установление взаимосвязи между структурой белка и его функциями. В настоящее время, в связи с определением нуклеотидной последовательности геномов ряда организмов, актуальной стала задача использования полученной при этом информации для детального понимания функционирования живой материи на молекулярном уровне.
В-третьих, поскольку биологическая функция белков определяется прежде всего их пространственным (трехмерным) строением, то задача выявления закономерностей фолдинга белка (т.е. складывания полипептидной цепи в функционально активную трехмерную структуру) является сейчас центральной. Понимание механизмов фолдинга также важно для биоинформатики и развития белковой инженерии.
Целью данной работы является обзор информационных технологий, компьютерных программ и баз данных, применяемых для анализа структуры и функций белков. Для этого были поставлены следующие задачи:
- ознакомление с литературными источниками для выявления основных направлений исследования белков с помощью средств информатики;
- проведение анализа последовательности природного белка разнообразными по назначению программами.
Глава 1. Обзор литературы
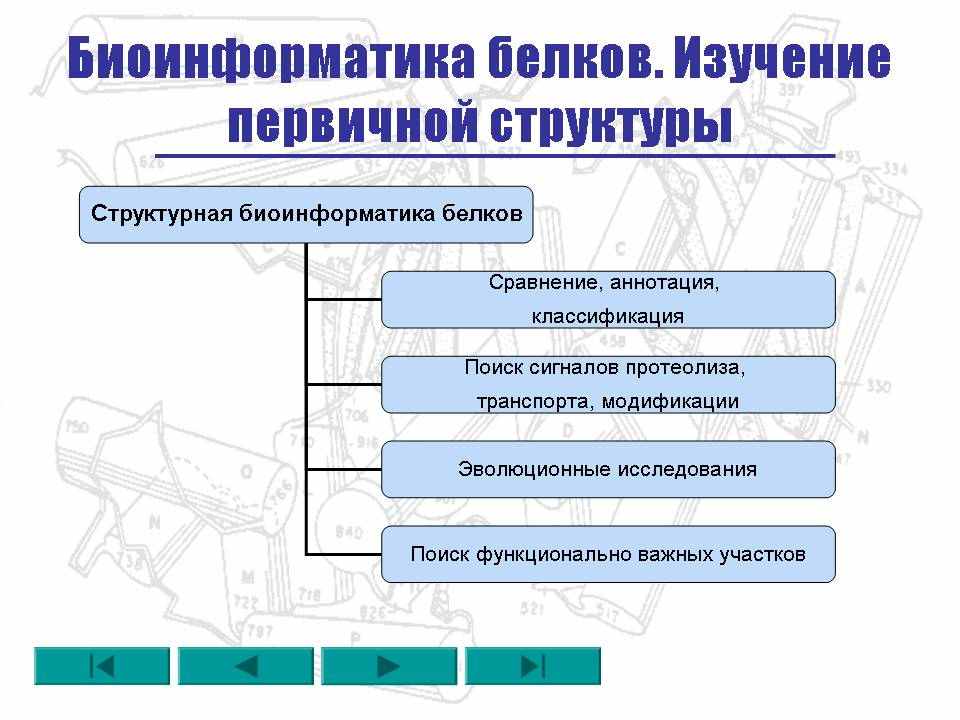
1.1 Изучение первичной структуры
Каждый белок, помимо своей уникальной последовательности аминокислот (первичная структура) , обладает ещё и уникальным способом укладки этой цепочки в пространстве (вторичная и третичная структуры). Задачу предсказания укладки по последовательности можно назвать важнейшей задачей биоинформатики, но это задача ещё слишком далека от своего решения. Поэтому структурная биоинформатика занимается анализом пространственных структур, уже определённых экспериментально, а также анализом первичной структуры белков [4].
Трёхмерных структур белков известно намного меньше, чем последовательностей, так как экспериментальные процедуры для определения структуры намного сложнее и дороже. Тем не менее, на начало 2007 года для анализа были доступны более 200 тысяч трёхмерных структур и несколько миллионов белковых последовательностей.
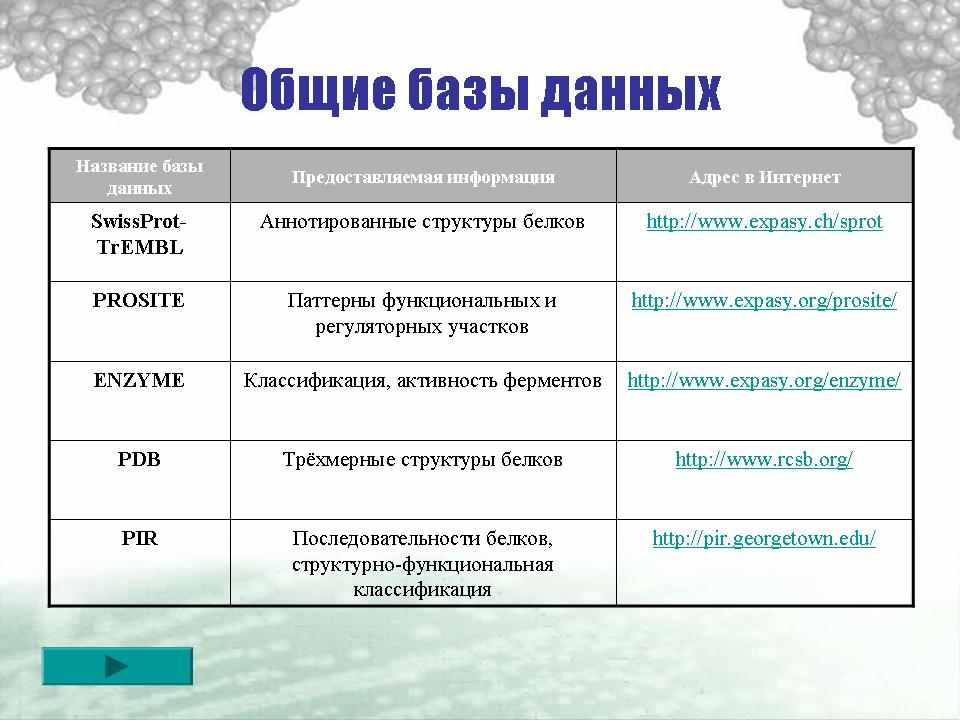
Существование огромного количества разнообразных белков привело к необходимости создания информационных массивов – баз данных , в которые заносились бы все известные о них сведения. Сейчас существует множество общих и специализированных баз данных, большинство которых предоставляет свободный доступ к разнообразным аминокислотным последовательностям, а также к удобным инструментам для обработки и анализа этой информации. Наиболее значительными базами данных на сегодня являются SwissProt-TrEMBL, PIR, PDB [4].
1.2 Моделирование трёхмерной структуры белков
Предсказание пространственной структуры белков по аминокислотной последовательности является одной из центральных задач компьютерной биологии.
Формально, зная взаимодействие между отдельными атомами в полипептидной цепи с известной первичной структурой и окружающими такую цепь молекулами растворителя, путем минимизации свободной энергии всей системы можно было бы найти искомую структуру. Даже для небольшого белка это задача поиска глобального минимума функции десятков тысяч переменных. Бесперспективность точного решения подобной задачи с помощью любого мыслимого суперкомпьютера вполне очевидна [6].
В настоящее время для решения проблемы фолдинга разработано большое число приближенных подходов. Один из наиболее эффективных - использование информации о гомологии, то есть о пространственной структуре белков, обладающих первичной структурой, близкой к исследуемому белку. Известная пространственная структура белка, гомологичного исследуемому, берется в качестве начального приближения, а затем производится ее уточнение [2]. Такие возможности предоставляют многие компьютерные инструменты для моделирования и анализа белковых структур, например SWISS-MODEL (ссылка скрыта), 3Djigsaw (ссылка скрыта) и другие.
Таким образом, компьютерный анализ белковых последовательностей и структур состоит из следующих основных элементов:
- предварительная аннотация по сходству и другим особенностям белковых последовательностей;
- сравнительный анализ структур родственных белков, классификация белков на основе их пространственной структуры;
- исследование регуляции транспорта или специфического протеолиза путём нахождения определённых аминокислотных сигналов;
- анализ и исследование эволюции белков, построение эволюционных схем.
- определение участков белковой молекулы, важных для той или иной функции данного белка;
- анализ структур комплексов двух или нескольких молекул белка, комплексов молекул белка с другими молекулами; предсказание воздействия молекул химических веществ (в частности, потенциальных лекарств) на молекулы белков;
- предсказание трёхмерной структуры белка по известной трёхмерной структуре белка с похожей последовательностью (в такой ситуации задача предсказания укладки часто возможна) .
1.3 Функциональная протеомика
Наличие в организме того или иного белка дает основание предполагать, что он обладает определенной функцией, а весь протеом служит для того, чтобы осуществлялась полноценная жизнедеятельность всего организма. Функциональная протеомика занимается определением функциональных свойств протеома, и решаемые ею задачи существенно сложнее, чем, например, определение белково-пептидных структур [4].
Очевидно, что функционирование протеома осуществляется в многокомпонентной среде, в которой присутствует множество молекул других химических классов – сахаров, липидов, простагландинов, различных ионов и многих других, включая молекулы воды. Белковые молекулы вступают в химические реакции с окружающими их молекулами или структурами, что в конечном итоге приводит к возникновению функциональных реакций не только на молекулярном уровне, но и на макроскопическом. Примерами таких процессов могут быть взаимодействие фермента с субстратом, антигена с антителом, пептидов с рецепторами, токсинов с ионными каналами. Для выявления механизмов этих процессов проводятся как экспериментальные исследования индивидуальных участников взаимодействия, однако более важным является изучение всей системы реакции определённого белка средствами биоинформатики.
Такие исследования позволяют выяснить принципы взаимодействия между белками, а также закономерности регуляции их работы. Функциональная протеомика ставит своей целью получить информацию о взаимодействии всех белков клетки и их влиянии на многие аспекты жизни клетки. Создание представлений об огромной сети взаимодействий белковых и других молекул в организме требует огромного труда и применения всех средств современной биоинформатики. По существу, создание таких представлений находится ещё в стадии развития .
Одним из первых успехов функциональной протеомики является создание карты метаболизма карбоновых кислот в Институте биохимии им. А.Н. Баха Российской академии наук. Эта карта представляет собой схему сети реакций с регулярным периодическим строением. Данная карта обладает прогностической силой, так как с её помощью было предсказано существование целого ряда неоткрытых ферментов, которые впоследствии были обнаружены экспериментально [4].
Глава 2. Методика исследования белковых последовательностей
2.1 Общие базы данных
Существование огромного количества разнообразных белков привело к необходимости создания информационных массивов – баз данных , в которые заносились бы все известные о них сведения. Сейчас существует множество общих и специализированных баз данных [1].
В общих базах содержатся сведения о всех известных белках живых организмов, т.е. о глобальном протеоме всего живого. Примером такой базы является SwissProt-TrEMBL (ссылка скрыта) , в которой на сегодняшний день содержатся структуры почти 300 000 белков, установленные аналитическими методами, и еще почти 10 млн структур, которые определены в результате трансляции с нуклеотидных последовательностей, и эти цифры постоянно увеличиваются [5].
База данных PIR (Protein Information Resource) (ссылка скрыта), являясь альтернативой SwissProt-TrEMBL, также осуществляет сбор данных о белках. Сейчас база PIR - продукт сотрудничества NBRF (США), MIPS (Германия) и JIPID (Japan International Protein Information Database). PIR предоставляет следующие возможности: банк последовательностей, структурно-функциональная классификация, аннотация и сравнение последовательностей, доступ к базам трёхмерных структур, и многие другие
С ресурсом SwissProt тесно связаны такие базы данных, как ENZYME и PROSITE. ENZYME (ссылка скрыта) представляет собой банк по белкам с каталитической активностью (ферментам) и содержит исчерпывающую информацию об месте в классификации, каталитической активности, кофакторах более чем 4000 различных ферментов. Также здесь присутствуют ссылки на структуры этих белков в SwissProt-TrEMBL [5].
База PROSITE (ссылка скрыта) содержит данные о различных паттернах (или мотивах) функциональных и регуляторных участков. С помощью этой коллекции можно определить семейство белков, к которому принадлежит новая последовательность, важный домен, который она содержит, классифицировать новый белок [5].
Широко признанной является ресурс PDB (Protein Data Bank) (ссылка скрыта) - коллекция экспериментально определенных трёхмерных структур биологических макромолекул. Начиная с 2002 года в ней хранятся только экспериментально определенные структуры (рентгеноструктурным, ЯМР и другими методами). Доступны также теоретические структуры, выделенные в отдельную подбазу, которая доступна для перекачки по FTP. На данный момент PDB содержит информацию о более чем 63 тысячах третичных структур белков и предоставляет богатые возможности для их анализа и сравнения.
В глобальном протеоме особое место занимают небольшие молекулы, содержащие не более 50 аминокислотных остатков и обладающие специфическим спектром функциональной активности - пептиды. Для них создан особый банк данных, который называется EROP-Moscow (ссылка скрыта). На сегодняшний день здесь содержится структура почти 6000 эндогенных регуляторных олигопептидов, выделенных из разнообразных организмов [4].
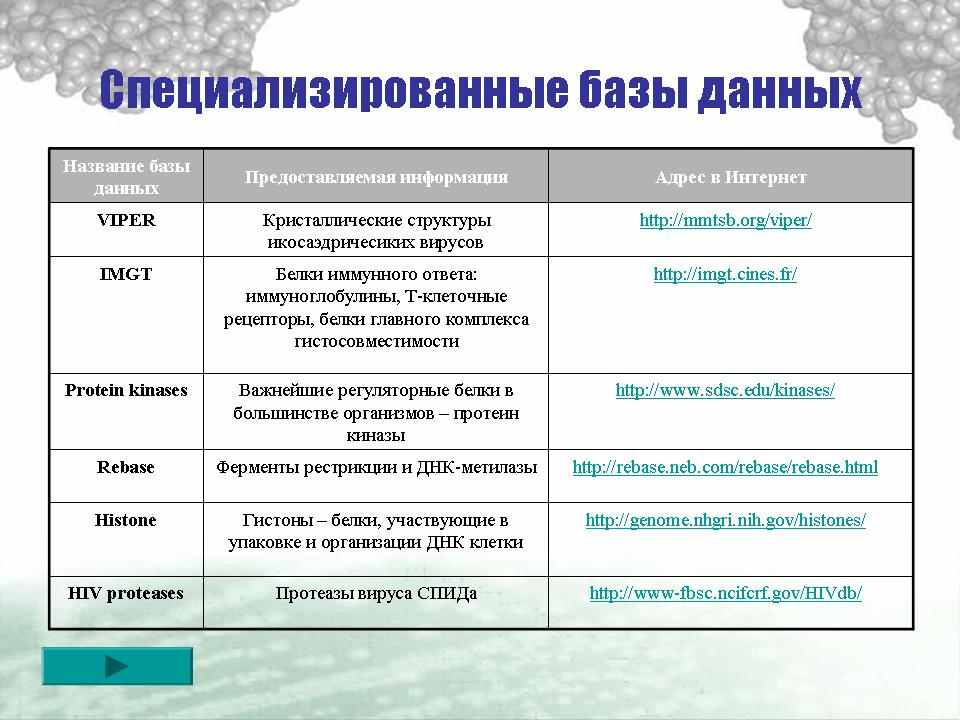
2.2 Специализированные базы данных
Специализированные базы данных собирают, аннотируют и систематизируют данные, сфокусированные на лишь определённых конкретных аспектах протеомики. Ниже представлены лишь некоторые примеры таких баз данных (табл. 1).
2.3 Сравнение последовательностей
Сравнение (согласование) нуклеотидных или аминокислотных последовательностей подразумевает последовательное сравнение звеньев биополимера, расположенных в соответствующих друг другу позициях в двух или более последовательностей. Это согласование подразумевает под собой не только сравнение, но и определение некоей количественной меры сходства последовательностей.
Таблица 1 – Примеры специализированных баз данных
| Название базы данных | Предоставляемая информация | Адрес в Интернет |
| VIPER | Кристаллические структуры икосаэдричесиких вирусов | ссылка скрыта |
| IMGT | Белки иммунного ответа: иммуноглобулины, Т-клеточные рецепторы, белки главного комплекса гистосовместимости | ссылка скрыта |
| Protein kinases | Важнейшие регуляторные белки в большинстве организмов – протеин киназы | ссылка скрыта |
| Rebase | Ферменты рестрикции и ДНК-метилазы | ссылка скрыта |
| Histone | Гистоны – белки, участвующие в упаковке и организации ДНК клетки | ссылка скрыта |
| HIV proteases | Протеазы вируса СПИДа | ссылка скрыта |
Сравнение последовательностей – важный инструмент для биоинформатики. Существует множество разнообразных алгоритмов для такого сопоставления. Основным из них является метод точечного графика (dotplot). Он позволяет охватить в единой картине не только общее сходство двух последовательностей, но также и любых других возможных соответствий. Другими производительными методами является использование различных математических матриц [2].
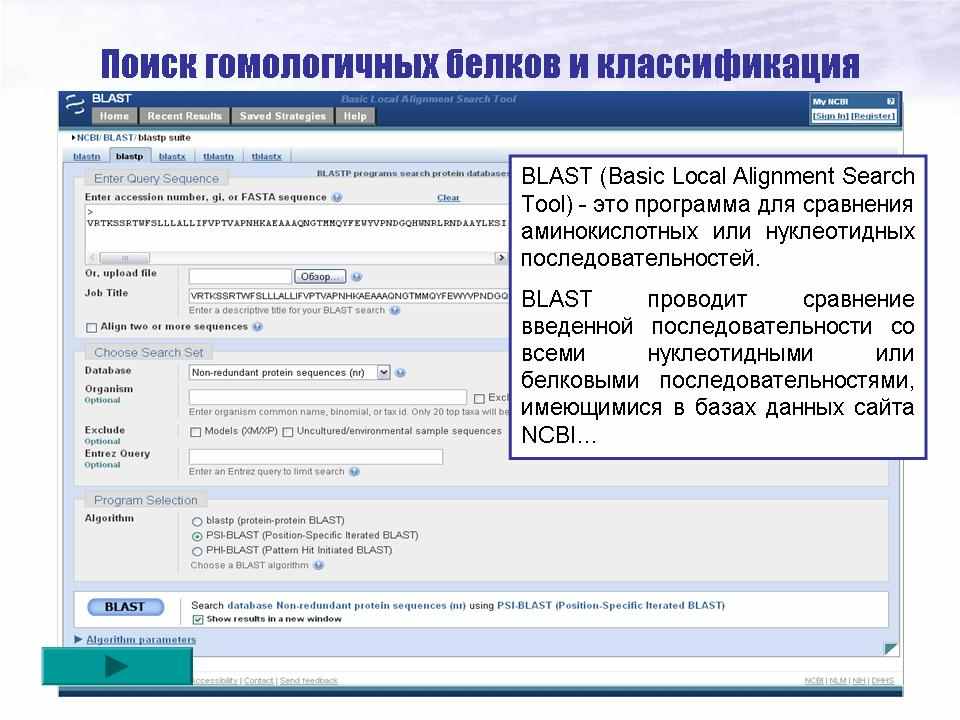
Для поиска схожих белков в огромных массивах баз данных известных белковых последовательностей на сегодня существуют 3 основных механизма: Profiles, PSI-BLAST и Hidden Markov Models (HMM) [2]. Из них выделяется PSI-BLAST, сочетающий высокую эффективность, быстроту и, что немаловажно, обладающий собственным удобным интерфейсом BLAST (ссылка скрыта).
2.4 Моделирование вторичной структуры
Как уже говорилось выше, предсказание третичных структур пока не достижимо для учёных. Предсказание вторичных структур, то есть структурных элементов третичной структуры - α-спиралей и β-слоёв, может быть осуществлено с помощью компьютерных программ. Предсказать вторичную структуру должно быть легче, так как нужно учитывать гораздо меньшее количество переменных взаимодействия между аминокислотами, и только в локальных участках, а не по всей молекуле. Кроме того, для прогнозирования третичной структуры разумным способом кажется моделирование элементов вторичной структуры, а затем сборка их в третичную [2].
На сегодняшний день важным шагом вперед в предсказании вторичной структуры стало создание алгоритмов, работающих по принципу нейронных сетей мозга человека (neural networks) [2]. Примерами программ, которые позволяют проводить такое моделирование, являются:
- Phyre Server (ссылка скрыта);
- Zhang-Server (ссылка скрыта);
- RAPTOR (ссылка скрыта);
- GORIV (ссылка скрыта).
Глава 3. Компьютерный анализ аминокислотной последовательности гена амилазы штамма Bacillus sp. 406
Амилолитические ферменты (или амилазы) катализируют гидролиз полимеров крахмала на небольшие углеводные фрагменты. Бактериальные α-амилазы, а особенно ферменты рода Bacillus, широко применяются в промышленных процессах благодаря высокой термостабильности и эффективной системе экспрессии у данных микроорганизмов. Экспериментально была определена нуклеотидная последовательность гена такого фермента, синтезируемого штаммом Bacillus sp. 406. Аминокислотная последовательность амилазы была определена по нуклеотидной последовательности, которая соответствует полипептидной цепи длиной 515 аминокислотных остатков. С помощью средств биоинформатики была предсказана информация о гомологии, сигнальных последовательностях, эволюционной принадлежности, свойствах и вторичной структуре данного белка.
3.1 Обнаружение гомологичных белков и классификация
Аминокислотная последовательность была сопоставлена с базой известных последовательностей амилолитических белков с помощью программы BLAST (Basic Local Alignment Search Tool) (ссылка скрыта). Это программа для логического сравнения как аминокислотных, так и нуклеотидных последовательностей. BLAST проводит сравнение введенной последовательности со всеми нуклеотидными или белковыми последовательностями, имеющимися в базах данных сайта NCBI, и выдаёт исчерпывающие результаты сравнения. BLAST используется для установки родственных связей, оценки функциональных особенностей последовательностей. Программа выдаёт результаты сравнения, а именно: список гомологичных белков в иерархическом порядке уменьшения схожести, ссылки на гомологичные последовательности с матрицей сравнения с исходной.

Рисунок 1 – Стартовый интерфейс программы BLAST.
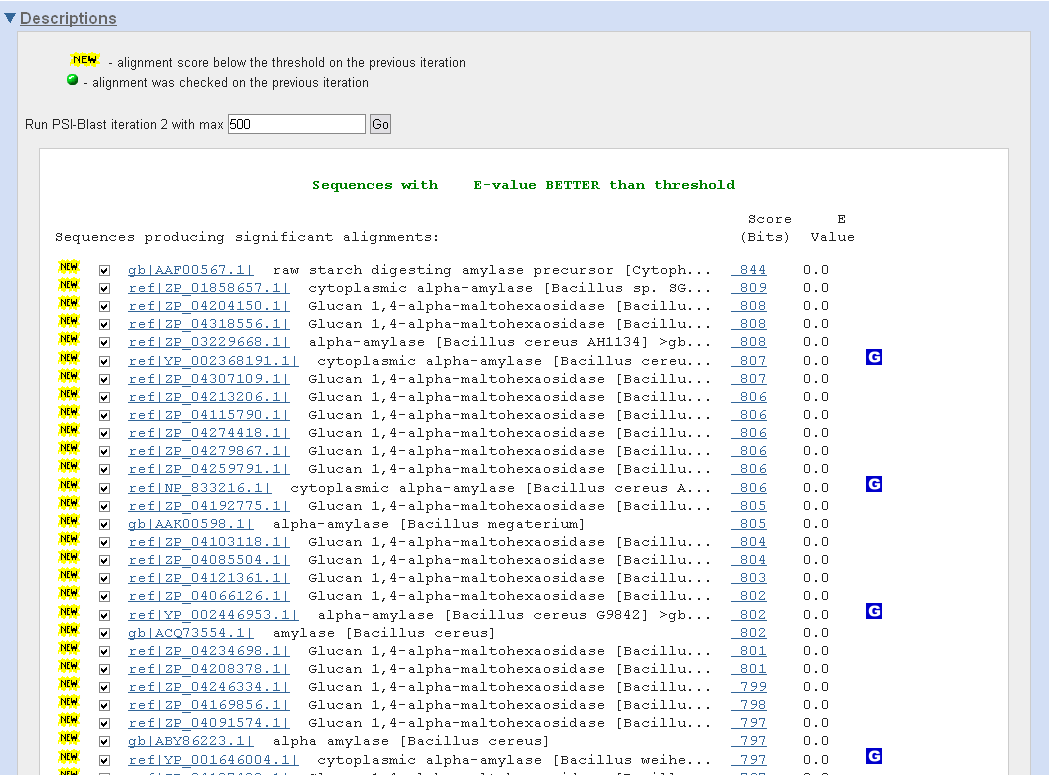
Рисунок 2 – Результаты сопоставления аминокислотных последовательностей в программе BLAST в кратком виде.

Рисунок 3 – Результаты сопоставления аминокислотных последовательностей в программе BLAST в расширенном виде.
Оказалось, что исследуемая последовательность проявляет наиболее высокую гомологию с α-амилазой Cytophaga sp.(идентичных аминокислот – 78 %, сходных – 88 %), α-амилазами Bacillus cereus, B. megaterium, B. weihenstephanensis, B. anthracis и B. thuringiensis (идентичных аминокислот – 74-76 %, сходных – 84-85 %).
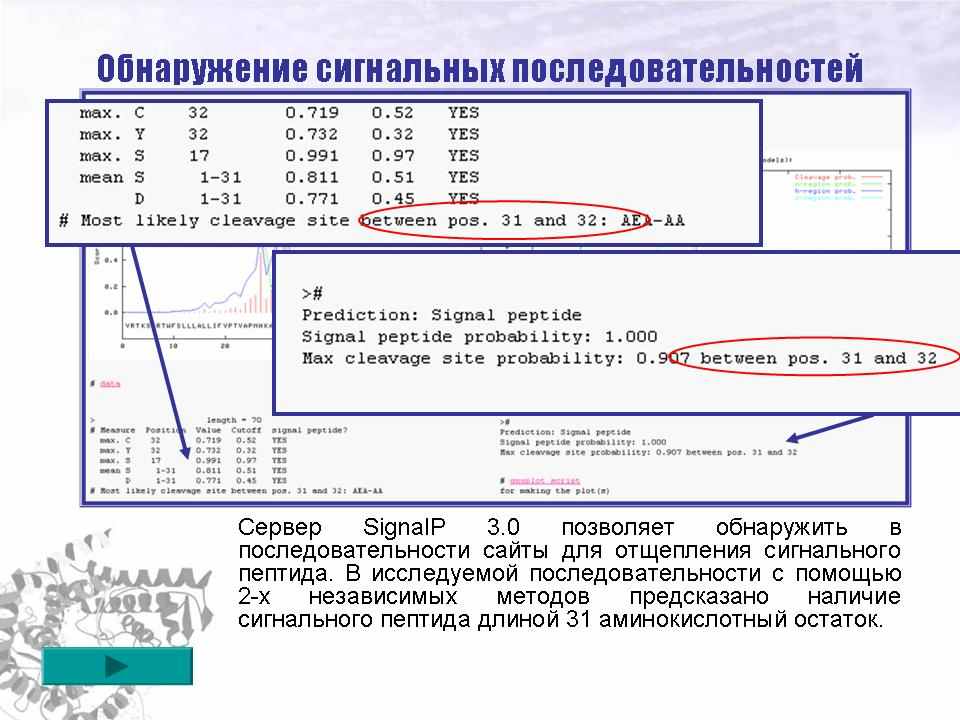
3.2 Обнаружение сигнальных последовательностей
Сервер SignalP 3.0 (ссылка скрыта) позволяет обнаружить в аминокислотной последовательности сайты для отщепления сигнального пептида, что позволяет говорить о характере транспорта и локализации белка. Алгоритм метода основан на использовании искусственных нейронных сетей и скрытых моделей Маркова, что увеличивает достоверность результата. В исследуемой последовательности с помощью этой программы была обнаружена область, соответствующая сигнальному пептиду длиной 31 аминокислотный остаток (рис. 4). Его расположение типично для сигнальных пептидов белков бактерий рода Bacillus.
3.3 Анализ физико-химических параметров белка
С помощью программы ProtParam (ссылка скрыта) на основании полученной аминокислотной последовательности были рассчитаны молекулярная масса и теоретическая pI амилазы Bacillus sp. 406. Для внутриклеточного предшественника фермента (с сигнальным пептидом) эти значения составляют 58,3 кДа и 6,26 соответственно. Для секретированного фермента (без сигнального пептида) эти значения равны 54,9 кДа и 5,93 соответственно. Кроме того, программа позволяет предсказать другие физико-химические параметры белка, например, состав аминокислотных остатков, атомов, коэффициент экстинкции, индексы нестабильности, гидрофобности.
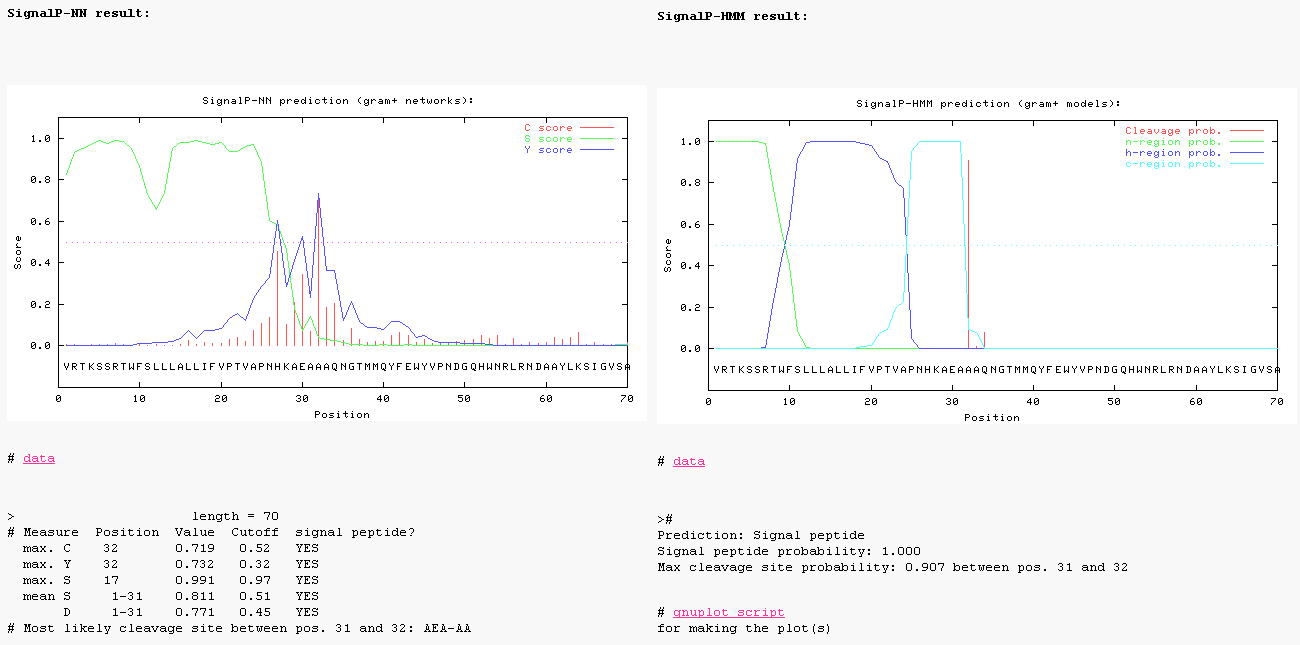
Рисунок 4 – Результаты предсказания положения сигнального пептида в программе SignalP по алгоритму нейронных цепей (слева) и скрытых моделей Маркова (справа).
3.4 Моделирование вторичной структуры
Предсказание вторичных структур, то есть структурных элементов третичной структуры - α-спиралей и β-слоёв, может быть осуществлено с помощью компьютерных программ, в отличие от третичной (пространственной) структуры всего белка. Принципом такого моделирования является предсказание взаимодействий между аминокислотами небольших участков всей последовательности белка, что программы выполняют с помощью разнообразных алгоритмов. В результате последовательность разбивается на линейный ряд участков, соответствующих одному из трёх элементов вторичной структуры - α-спирали, β-слою или неструктурированному участку.
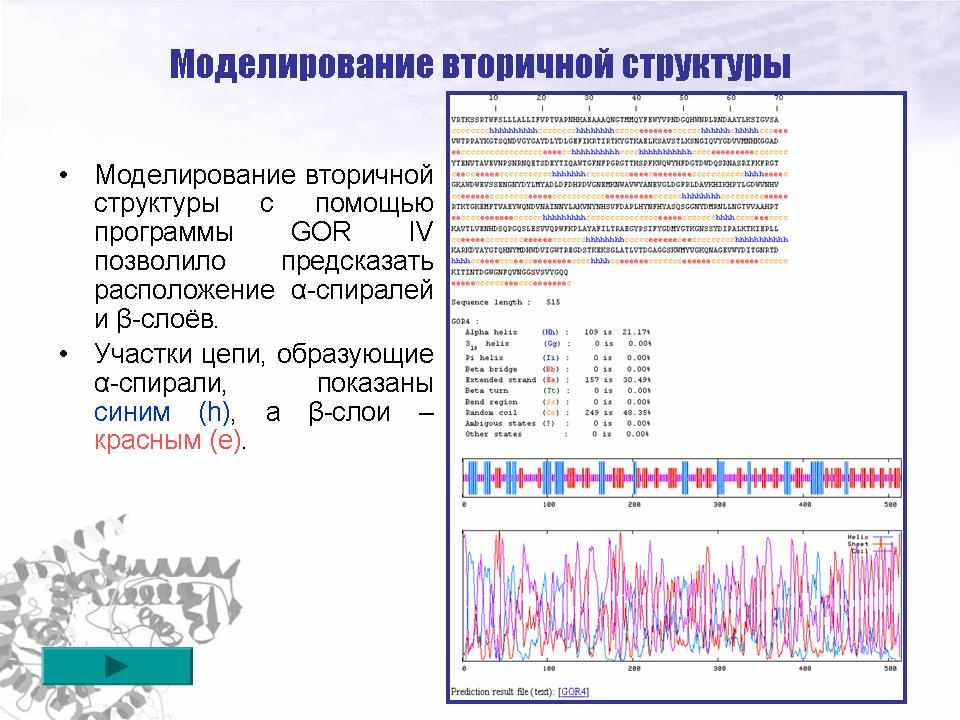
Моделирование вторичной структуры с помощью программ Phyre Server (ссылка скрыта) и GOR IV (ссылка скрыта) позволило предсказать расположение α-спиралей и β-слоёв. Phyre Server выдаёт данные в текстовом формате, GOR IV – в текстовом и удобном графическом (рис. 5).

Рисунок 5 – Результаты моделирования вторичной структуры на основе последовательности амилазы Bacillus sp. 406 в программе GOR IV.
Результаты моделирования этими программами были практически идентичными и показали, что центральный домен белка содержит восемь β-слоёв, чередующихся с восьмью α-спиралями. Эта структура схожа с (β/α)8 TIM цилиндром, формирующим ядро известных амилаз и формирующих каталитический центр фермента. С двух сторон от центрального домена имеются 2 участка, богатые β-слоями. Они также соответствуют присутствующим с амилазах доменам В и С. Таким образом, предположительная вторичная структура амилазы Bacillus sp. 406 типична для α-амилаз бактерий рода Bacillus.
3.5 Эволюционный анализ
Программа MEGA 4.0 (ссылка скрыта) включает в себя широкий диапазон статистических и вычислительных методов для сравнительного анализа последовательностей. Она позволяет анализировать и сопоставлять как нуклеотидные, так и аминокислотные последовательности (рис. 6), осуществлять поиск гомологии в Интернет-базах данных, имеет гибкие возможности вывода аннотированных данных о последовательностях [8].
Рисунок 6 – Инструмент анализа аминокислотных последовательностей в программе MEGA 4.0.
Важной особенностью программы является возможность проводить расчёт эволюционной дистанции с разнообразными настройками, генерировать филогенетические деревья и редактировать их.
С помощью MEGA 4.0 на основе аминокислотных последовательностей α-амилаз, близких по гомологии с исследуемым ферментом, была определена предположительная эволюционная принадлежность амилазы Bacillus sp. 406 (рис. 7).
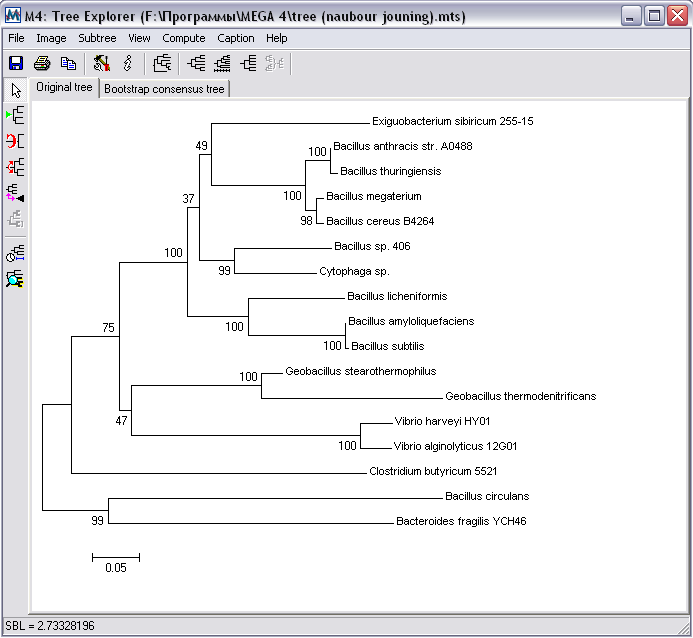
Рисунок 7 – Филогенетическое дерево на основе аминокислотных последовательностей α-амилаз различных грамположительных организмов, построенное в программе MEGA 4.0.
Заключение
В последние десятилетия развитие информационных технологий начало затрагивать все сферы жизни человека. Одно из важнейших мест в этом списке занимает наука. Если два столетия назад биология сводилась лишь к описательным приёмам, то сейчас перед нами сложная разнообразная наука, вооружённая математическим аппаратом и информационными технологиями. Даже родоначальники генетики и синтетической теории эволюции, поставившие биологию на ноги математики, не могли себе представить, что в XXI веке компьютеры позволят человеку оперировать немыслимыми объёмами информации (последовательности генов, белков, целые геномы), моделировать сложнейшие комплексные процессы (сворачивание белка, моделирование эволюционных процессов), анализировать и систематизировать самые разнообразные данные. Биоинформатика имеет и существенную практическую ценность, например, в фармацевтике при поиске новых лекарственных препаратов.
Биоинформатика является очень перспективным направлением биологии. Сегодня учреждаются специальные компании и научные центры, работающие в сфере биоинформатики. Высшие учебные заведения создают факультеты, подготавливающие высококвалифицированных специалистов в этой дисциплине. Специализированное программное обеспечение в лаборатории стало столь же необходимым, как и лабораторное оборудование.
Традиционно к задачам биоинформатики относятся: статистический анализ последовательностей, предсказание функции по последовательности, анализ пространственной структуры белков и нуклеиновых кислот. Однако, в последние годы возник ряд новых задач, связанных с прогрессом в области автоматизации многих экспериментальных процедур, анализа белок-белковых взаимодействий, исследования работы генов в различных условиях. При этом не только возникает необходимость создавать новые алгоритмы, но и происходит распространение подходов биоинформатики на смежные области, например, популяционную и медицинскую генетику. Таким образом, роль биоинформатики перестала сводиться к обслуживанию экспериментальной науки - у нее появились свои собственные задачи. Это говорит о том, что эта область знаний превратилась в самостоятельную науку.
Предметный указатель
B
Bacillus 16, 19, 21, 22
BLAST 14, 16
S
SwissProt 8, 9, 12
а
алгоритм 14, 15, 19, 20, 24
амилаза 16
аминокислотная последовательность 8, 12, 16
б
база данных 8, 12, 13, 16
биоинформатика 5, 8, 24
в
вторичная структура 15, 20, 21
м
моделирование 6, 9, 15, 20, 21
н
нейронные сети 15, 19
нуклеотидная последовательность 5, 16
п
программа 16, 19, 21, 22
протеомика 5, 6, 10, 11
с
сравнение последовательностей 9, 13, 16
т
трёхмерная структура 8, 12, 13
ф
фолдинг 6, 9, 10, 15
Список Иллюстраций
Рисунок 1 – Стартовый интерфейс программы BLAST. 17
Рисунок 2 – Результаты сопоставления аминокислотных последовательностей в программе BLAST в кратком виде. 18
Рисунок 3 – Результаты сопоставления аминокислотных последовательностей в программе BLAST в расширенном виде. 18
Рисунок 4 – Результаты предсказания положения сигнального пептида в программе SignalP по алгоритму нейронных цепей (слева) и скрытых моделей Маркова (справа). 20
Рисунок 5 – Результаты моделирования вторичной структуры на основе последовательности амилазы Bacillus sp. 406 в программе GOR IV. 21
Рисунок 6 – Инструмент анализа аминокислотных последовательностей в программе MEGA 4.0. 22
Рисунок 7 – Филогенетическое дерево на основе аминокислотных последовательностей α-амилаз различных грамположительных организмов, построенное в программе MEGA 4.0. 23
Список использованной литературы
- Чемерис А.В., Ахунов Э.Д., Вахитов В.А. Секвенирование ДНК. - М.: Наука, 1999. - 429 с.
- Lesk A. M. Introduction to Bioinformatics. - Oxford University Press, USA, 2002. - 320 р.
- ссылка скрыта
- ссылка скрыта
- ссылка скрыта
- su.ru/Site_files/about_fbb/about_bioinformatic.php">
- ссылка скрыта
- ссылка скрыта
- ссылка скрыта
- ссылка скрыта
- ссылка скрыта
Интернет-ресурсы в предметной области
ссылка скрыта
«Биомолекула» — это интернет-издание, посвященное молекулярным основам современной биологии и практическим применением научных достижений в медицине и биотехнологии. Публикуемые материалы носят научно-популярный характер, информация излагается доступным языком для любого интересующегося читателя, не теряя при этом своей научной достоверности.
ссылка скрыта
Проблемы эволюции. Сайт предназначен для всех, кто интересуется эволюцией. Здесь можно найти богатейшую библиотеку популярных и научных трудов по эволюции (более 600 работ), обзоры и палеонтологические иллюстрации. Имеется развитый форум.
ссылка скрыта
ExPASy Proteomics Server. Сервер протеомики Швейцарского Института Биоинформатики. Содержит множество баз данных по разным областям исследований белков, а также богатейшую коллекцию инструментов для анализа белковых последовательностей и структур.
ссылка скрыта
Биология человека. Сайт представляет собой своего рода энциклопедию по данным, касающимся физиологии, цитологии, молекулярных и генетических аспектов человека и биологической жизни. Ресурс имеет удобную систему ссылок, поиск и форум.
ссылка скрыта
Объединённый центр вычислительной биологии и биоинформатики. Сайт института математических проблем биологии РАН. Содержит информацию о работе института, его проектах. Сайт особенно полезен большой и структурированной коллекцией ссылок на самые разнообразные ресурсы, посвящённые биологии и биоинформатике (сайты организаций, крупнейшие центров биоинформатики, биоинформационных веб-ресурсов, источники книг, справочников, журналов, банков данных).
ссылка скрыта
Классическая и молекулярная биология. Этот популярный ресурс содержит самую разнообразную информацию для биологов, особенно для генетиков, молекулярных биологов, биотехнологов и биохимиков. Сайт содержит руководства и протоколы экспериментальных методов исследования, прописи приготовления реактивов и растворов, постоянно обновляемую информацию о свежих публикациях и научных мероприятиях. Имеется множество ссылок на сайты биологических научных центров, фирм, ресурсов, а также форум.
ссылка скрыта
National Center for Biotechnology Information (NCBI) - крупнейшая биологическая база данных. NCBI предоставляет мощные системы обработки и представления этих данных. Содержит следующие базы данных и программы для поиска: GenBankRefSeq, DbSNP, UniGene, OMIM, Genomic Bi-ology, Entrez, Entrez Genomes, BLAST, базы аминокислотных последовательностей, базы нуклеотидных последовательностей. Имеются ссылки на базы данных научных журналов.
ссылка скрыта
PubMed - информационный ресурс Национального Института Здравоохранения США. Cодержит огромную базу статей из научных журналов медицинской и естественнонаучной направленности. Размещены ссылки на полные тексты статей и другие связанные ресурсы, имеется удобный механизм поиска.
ссылка скрыта
"Вся биология" - это научно-образовательный проект, посвящённый биологии и родственным наукам. Большой информационный ресурс, главная цель которого: предоставление информации по всем разделам биологии в максимально доступной форме для обычного читателя. На портале можно почитать новости науки, обзоры, пообщаться на форуме.
ссылка скрыта
Сайт содержит множество on-line программ для анализа генетической и молекулярно-биологической информации.
ссылка скрыта
Сайт Высшей аттестационной комиссии Республики Беларусь. Содержит материалы и нормативную документацию, касающиеся подготовки научных кадров, присуждения ученых степеней и званий, краткие паспорта специальностей и программы кандидатских экзаменов по специальности, бланки различных документов.
Действующий личный сайт в Интернет
ссылка скрыта
Граф научных интересов
Аспиранта Качана А. В. биологический факультет
Специальность “Биология (биотехнология)”
| Смежные специальности | Основная специальность | ||||||||
|
| ||||||||
| Сопутствующие специальности | |||||||||
|
Презентация
выполненая в Power Point, находится по следующему адресу:
ссылка скрыта
Список литературы по выпускной работе
- Кишик А. Эффективный самоучитель Excel 2003 - СПб.: Питер, 2005. – 247 c.
- Спека М.В. Презентации MS Power Point 2003: самоучитель. - Диалектика, 2004. – 368 с.
- Хэлворсон М., Янг М. Эффективная работа: Office XP. - СПб.: Питер, 2004. —1072 с.
- Чемерис А.В., Ахунов Э.Д., Вахитов В.А. Секвенирование ДНК. - М.: Наука, 1999. - 429 с.
- Lesk A. M. Introduction to Bioinformatics. - Oxford University Press, USA, 2002. - 320 р.
- ссылка скрыта
- ссылка скрыта
- ссылка скрыта
- su.ru/Site_files/about_fbb/about_bioinformatic.php">
- ok.ru/html/td.php
- ссылка скрыта
- ok.ru/html/td.php
Приложение
Приложение