
Разработка научных основ, промышленная реализация и развитие сырьевой базы каталитических процессов получения синтетических олигоолефиновых масел на основе нефтяного и растительного сырья 02. 00. 06 высокомолекулярные соединения
| Вид материала | Автореферат диссертации |
СодержаниеНаучные основы новых селективных Селективный каталитический процесс получения бутена-1 методом димеризации этилена. |
- Носители противоопухолевых препаратов на основе синтетических полипептидов 02. 00., 548.13kb.
- «Кинетика и механизм реакции поликонденсации аминокислот» 02. 00. 04 физическая химия, 332.67kb.
- Математическое моделирование процессов в тепловых микросенсорах, 21.43kb.
- Биологически активные вещества каллусной ткани наперстянки пурпурной смольникова, 111.89kb.
- Стандартизация и контроль качества лекарственного растительного сырья стандартизация, 615.33kb.
- Стандартизация и контроль качества лекарственного растительного сырья стандартизация, 613.27kb.
- Тверской государственный технический университет реферат на тему, 430.37kb.
- Задание на проектирование Рассчитать оборудование для стадии выщелачивания исходного, 48.05kb.
- Агранович б. Л. Козлов, 1002.27kb.
- О чем рассказал заместитель министра?, 61.71kb.
НАУЧНЫЕ ОСНОВЫ НОВЫХ СЕЛЕКТИВНЫХ
КАТАЛИТИЧЕСКИХ ПРОЦЕССОВ ПОЛУЧЕНИЯ
ИНДИВИДУАЛЬНЫХ ВЫСШИХ АЛЬФА-, «ВНУТРЕННИХ-» И
ИЗООЛЕФИНОВ С4, С6, С8, С10, С12, С14
Целью и основными задачами рассматриваемых в настоящей главе этапа исследований и разработок являлось «создание фундаментальных научных основ и технологического оформления селективных каталитических процессов получения индивидуальных высших олефинов».
Многие олефины обычно получают в виде смеси гомологов в каталитических процессах статистической олигомеризации этилена, из которой обычно выделяют необходимый потребителю конкретный индивидуальный олефин. Теоретический анализ показал, что в процессах статистической олигомеризации, которая представляет собой частный случай полимеризационных процессов, образуются смеси гомологов линейных альфа-олефинов (ЛАО). Селективное получение индивидуальных высших олефинов с селективностью S ³ 95 мас. % невозможно.
Главными отличительными особенностями разрабатываемых процессов, которые обеспечивают существенные преимущества их перед существующими аналогами, являются: 1) возможность селективного получения индивидуальных высших олефинов из доступного ограниченно потребляемого и альтернативного сырья; 2) осуществление их в мягких технологически благоприятных условиях; 3) создание производства базисного сырья для многих многотоннажных процессов получения нефтехимических и полимерных продуктов.
Во всех случаях фундаментальные научные исследования включали разработку оригинальных высокоактивных и высокоселективных катализаторов, изучение характера влияния различных факторов на кинетические закономерности и селективность процессов, а также на характеристики высших олефинов. Это позволило выявить оптимальные условия изучаемых процессов и получить информацию об их механизмах. На основе совокупности результатов выполненных исследований были разработаны принципиальные технологические схемы разрабатываемых процессов.
Селективный каталитический процесс получения бутена-1 методом димеризации этилена. Обычно бутен-1 получают путем димеризации этилена под действием системы Ti(OR)4-AlR3 в среде углеводородных растворителей. Из-за бицентрового характера упомянутой каталитической системы в этом процессе наряду с бутеном-1 образуется значительное количество полиэтилена.
В результате изучения характера влияния природы среды на процессы регулируемой полимеризации этилена, природы активных центров и стадийного механизма димеризации этилена в бутен-1 найден комплекс новых решений, позволивших полностью устранить полимерообразование, повысить селективность процесса (за счет устранения полимерообразования и уменьшения выхода С6 -С8 олефинов), а также упростить его технологическое оформление. Основные усовершенствования известного процесса достигнуты за счет осуществления димеризации этилена в бутен-1 в среде простых эфиров. При этом выполнены следующие исследования:
1) выяснен характер влияния различных факторов на кинетические закономерности и селективность процесса димеризации этилена в бутен-1 под действием системы Тi(O н-C4H9)4 – AlR3 в среде простых эфиров (диэтилового, дибутилового, метилфенилового, этилфенилового, метилтретбутилового и др.);
2) изучена димеризация этилена под действием системы Тi(O н- C4H9)4 – Al(С2H5)3 в среде диэтилового эфира в присутствии водорода;
3) выяснены особенности взаимодействия Тi(O н-C4H9 )4 с AlR3 в среде кислород- и азотсодержащих растворителей:
3.1) показано, что исходные компоненты систем Тi(O н-C4H9)4 – AlR3 при температурах до 100оС в среде простых эфиров не реагируют с этиленом;
3.2) определен выход и состав газообразных продуктов реакций Тi(O н-C4H9)4 с AlR3 в среде простых эфиров;
3.3) методом ЭПР изучена кинетика накопления и расходования парамагнитных продуктов восстановления Тi(O н-C4H9)4 в системах Тi(O н-C4H9)4 – AlR3 в интермедиаты Ti(III) и Ti(I) в среде кислород- и азотсодержащих растворителей. Однозначно доказано, что комплексы Ti(III) включают фрагменты алюминийорганического соединения и не проявляют каталитической активности в процессе димеризации этилена в бутен-1;
3.4) показано, что ионные стадии не влияют ни на реакции в каталитических системах Тi(O н-C4H9)4 – AlR3, ни на процесс димеризации этилена под действием этих систем в среде простых эфиров.
Доказано, что:
4) основой активных центров димеризации этилена являются эфирные комплексы одновалентного титана, которые образуются в результате глубокого восстановления титана в системах Тi(O н-C4H9)4 – AlR3 алюминийорганическими соединениями;
5) одновалентный титан содержит не спаренный электрон и является титан центрированным радикалом;
6) активный центр димеризации не содержит фрагментов алюминийорганического сокатализатора;
7) в комплексах одновалентного титана имеется большое число легко освобождаемых под действием этилена координационных мест;
8) первая стадия процесса димеризации включает координацию двух молекул этилена на атоме Ti(I);
9) предложен возможный стадийный механизм димеризации этилена (схема 1), в соответствии с которым вслед за образованием комплексов Ti(I) ∙ 2 С2Н4 происходит окислительное присоединение двух молекул этилена к Ti(I) с образованием пятичленного титаноцикла TiC4H8 и с повышением степени окисления титана до трех;
10) показано, что последующий распад пятичленного титаноцикла с двухкратным гидридным переносом приводит к образованию бутена-1 и к восстановле-
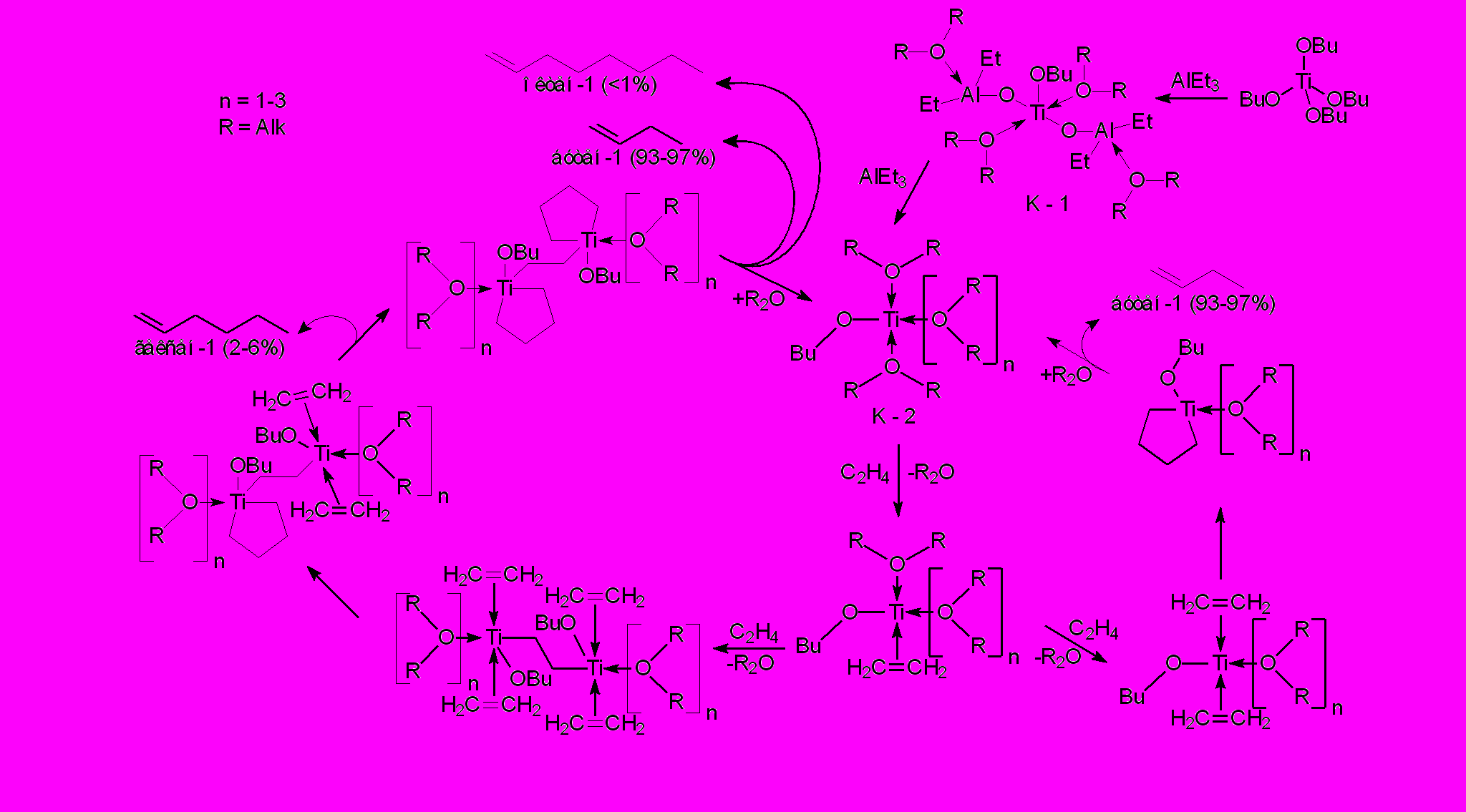
Схема 1. Механизм димеризации этилена в бутен-1
нию Ti(III) до Ti(I) с регенерацией активного центра в каждом каталитическом цикле. Лимитирующей стадией этого процесса является гидридный перенос. Это следует из наличия кинетического изотопного эффекта при сопоставлении кинетических закономерностей димеризации этилена и тетрадейтероэтилена;
11) экспериментально, методом встречного синтеза соединения титана с пятичленным титаноциклом, подтверждено, что стадии 9 и 10 являются обратимыми.
Такой специфический механизм процесса димеризации обеспечивает возможность функционирования каталитических систем Тi(O н-C4H9)4 – AlR3 в высоко сольватирующих и высокополярных средах (бензол, толуол, хлорбензол, хлористый этил, простые эфиры) при температурах 40-100оС и давлениях этилена 0.3-15 (преимущественно до 8) ат с выходом до 20 кг бутена-1 в расчете на один грамм Тi (т.е. с образованием свыше 15000 молей индивидуального бутена-1 в расчете на один моль Тi(O н-C4H9)4) в каталитической системе Тi(O н-C4H9)4 – AlR3.
Перечисленные признаки проявляются и в случае селективных процессов тримеризации этилена в гексен-1, тетрамеризации этилена в октен-1, содимеризации этилена с диенами и полимеризации ацетилена под действием парамагнитных комплексов Cr, Fe и Ni в низких степенях окисления.
Рассмотренные выше признаки селективных процессов ди-, три-, тетрамеризации и содимеризации этилена с диенами позволяют утверждать, что открыто новое направление катализа полимеризационных процессов.
Научные основы селективного процесса получения децена-5, додецена-6 и тетрадецена-7 методом метатезиса гексена-1, смесей гексена-1 с октеном-1 и октена-1, соответственно. Идеальным сырьем для получения ПАОМ (с точки зрения обеспечения всего комплекса физико-химических и потребительских свойств) является децен-1. Ресурсы его крайне ограничены.
В результате выполненных фундаментальных исследований по разработке научных основ технологии получения ПАОМ путем катионной олигомеризации децена-1 обнаружены явления, которые позволяют существенно расширить сырьевую базу получения деценовых ПАОМ за счет вовлечения в процесс менее потребляемого, а потому и менее дорогостоящего гексена-1:
установлено, что актом роста цепи в процессе катионной олигомеризации децена-1 под действием разработанных катионных катализаторов олигомеризации предшествует изомеризация децена-1 в смесь позиционных и геометрических изомеров децена-1, которые далее соолигомеризуются с деценом-1 в олигодецены;
- показано, что смеси цис- и транс- деценов-2, -3, -4 и –5 с высокой скоростью олигомеризуются под действием разработанных катионных катализаторов. Использование цис- и транс-деценов-5 в качестве исходного сырья для получения ПАОМ благоприятно влияет на фракционный состав продуктов олигомеризации (в сторону увеличения доли тримеров децена) и позволяет получать ди- и тримеры деценов с более низкими температурами застывания (до –90 и –750С, соответственно), чем в случае аналогичных продуктов, получаемых на основе децена-1.
Возможность переработки гексена-1 в децен-5 возникла после открытия Элеутерио в 1957 г. каталитической реакции метатезиса ЛАО по следующей формальной схеме:
2 R-CH=CH2 ® R-CH=CH-R + CH2=CH2
В процессах метатезиса используют как растворимые, так и гетеро-генизированные катализаторы на основе различных соединений Mo, W и Re.
С учетом выше сказанного разработаны оригинальные доступные нанесенные катализаторы - WCl6/SiO2-MeR4; MoO3/SiO2 или MoO3/Al2O3-RnAlCl3-n, применение которых позволило: 1) повысить стабильность катализатора в процессе приготовления, хранения, применения; 2) использовать катализатор без реактивации и активации его кислотами Льюиса при метатезисе нескольких порций гексена-1; 3) осуществить метатезис гексена-1 в массе; 4) осуществлять многократную окислительную реактивацию оксидных катализаторов и упростить технологию их выделения.
С целью выбора оптимальных условий метатезиса гексена-1 и получения информации о механизме процесса изучено влияние различных факторов на кинетику выделения и состав газообразных продуктов, образующихся в процессе диспропорционирования гексена-1 под действием систем MoO3/SiO2-АОС, MoO3/Al2O3-АОС при низких температурах (20-100оС) и низком содержании МоО3 на поверхности частиц SiO2 (1.08-4.0 мас. %). Оптимальная концентрация катализатора MoO3/SiO2 (Al/Mo=2.8) при этом равняется 40-50 г/л.
При постоянной концентрации АОС и прочих других равных условиях активность системы MoO3/SiO2 - АОС в процессе метатезиса гексена-1 в зависимости от природы RnAlCl3-n уменьшается в следующем ряду: Al(CH3)3 ³ Al(C2H5)3 > Al(изо-C4H9)3 > (CH3)2AlCl > (C2H5)2AlCl. Зависимость максимальной скорости реакции, эффективности катализатора и предельной конверсии гексена-1 на катализа-
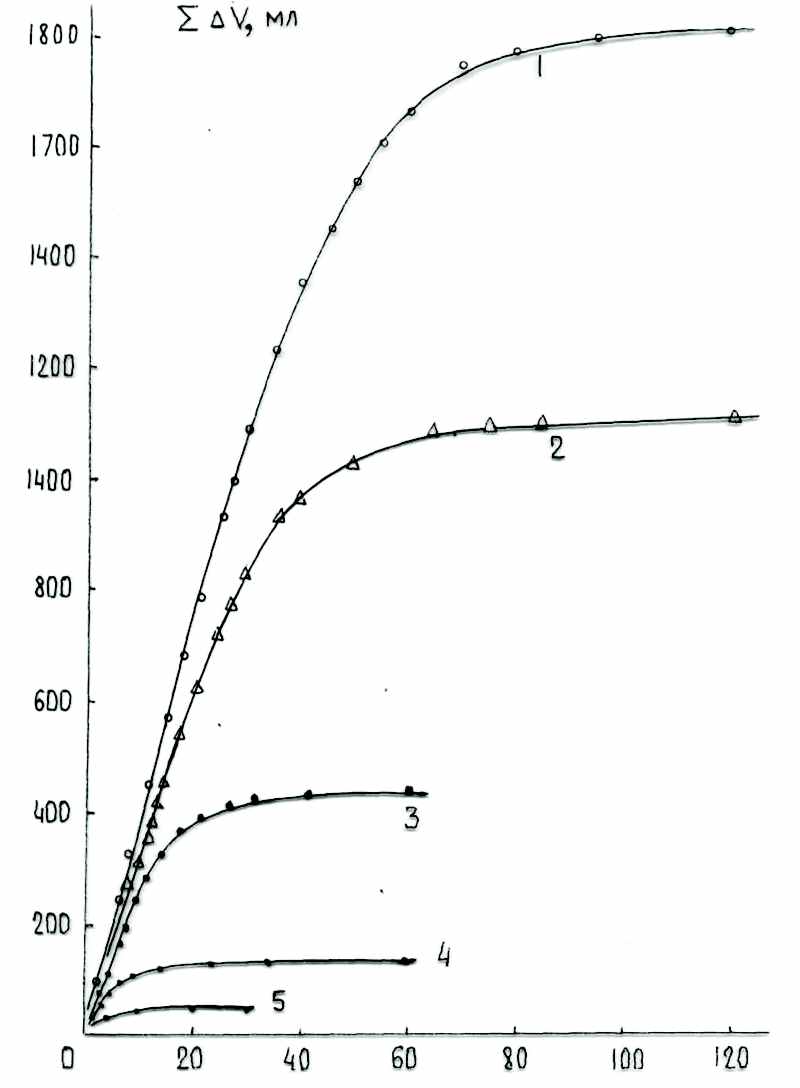 | 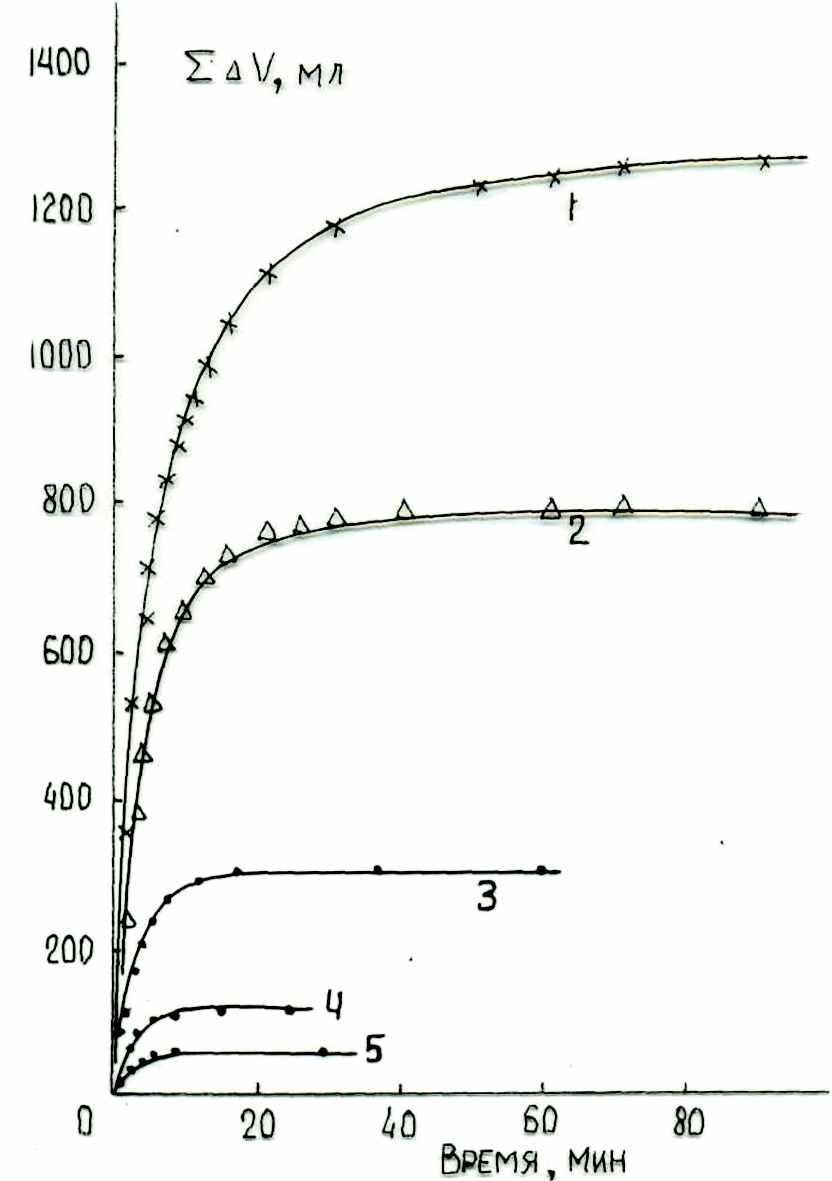 |
| Рис. 4. Влияние концентрации гексена-1 на кинетику выделения газообразных продуктов при диспропорционировании его под действием системы МоОз (1.61 мac.%)/SiO2 С-3 (г < 0.25 мм) при 293 °К; MoO3/SiO2 - 3.5 г; Мо - 0.0382 г - 0.4 ммоль; А1(СН3)3 - 1.19 ммоль (0.04 моль/л); А1/Мо - 3.0. № гексан, мл гексен-1, мл См, моль/л 1 0.0 30.0 8.00 2 10.0 18.0 5.35 3 20.0 10.0 2.67 4 25.0 5.0 1.33 5 27.5 0.5 0.66 | Рис. 5. Влияние концентрации децена-1 на кинетику выделения газообразных продуктов при диспропорционировании его под действием системы MoO3/SiO2 С-3 (г < 0.25 мм) - А1(СН3)з при 333 °К. Мо - 1.09 мас.%; (0.0382 г = 0.4 ммоль); MoO3/SiO2 - 3.5 г; А1(СН3)з -1.19 ммоль; А1/Мо = 3.0. № декан, мл децен-1, мл См, моль/л 1 3.1 30.0 5.30 2 13.1 20.0 3.54 3 23.1 10.0 1.77 4 28.1 5.0 0.88 5 30.6 2.5 0.44 |
торах рассматриваемого типа от концентрации АОС имеет экстремальный характер. Это свидетельствует о том, что АОС участвуют не только в образовании активных центров метатезиса, но и в их дезактивации. Селективность катализатора MoO3/SiO2 - АlR3 практически не зависит от концентрации АОС.
Детально изучено влияние концентрации гексена-1 на все характеристики его метатезиса (рис. 4, 5). Максимальная скорость монотонно возрастает с повышением концентраций гексена-1 и децена-1, соответственно. Формальный порядок ре-
акции по гексену-1 зависит от природы носителя и температуры: 0.35 (MoO3/SiO2 С-3 + Al(CH3)3+ гексен-1 при 20 оС), 0.87 (MoO3/SiO2 C-3 + Al(CH3)3 + децен-1 при 60 оС) и 0.45 (MoO3/SiO2 КСМ + Al(CH3)3 + гексен-1 при 20 оС). Эти наблюдения можно объяснить тем, что метатезис гексена-1 включает две стадии - обратимую координацию олефина на атоме Мо в активном центре и собственно метатезис:
M
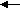
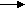 o = CH-R+ CH2 =CH-R [Z] Mo=CH2 +RCH=CHR (децен-5)
o = CH-R+ CH2 =CH-R [Z] Mo=CH2 +RCH=CHR (децен-5)k1 k2
M
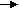
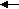
 o = CH2+ CH2=CH-R [Z1] Mo=CH-R + C2H4 ↑,
o = CH2+ CH2=CH-R [Z1] Mo=CH-R + C2H4 ↑, k-1
где Z и Z1 – промежуточные комплексы металлкарбен-гидридных активных центров метатезиса с гексеном-1, а R - C4H9
Mo = CH2 - C4H9 + CH2 = CH2 - C4H9 ® Mo= CH2 + C4H9CH = CHC4H9 (децен-5)
Анализ этой модели в квазистационарном приближении позволил получить зависимость начальной скорости выделения этилена (Wo) от концентрации альфа-
олефина (М):
1/Wo = 1/k2∙Xo + (k-1+k2)/k1k2Xo∙M,
где Хо – концентрация активных центров, включающих фрагмент Мо=СН2
Экспериментальные данные о зависимости (Wo) от (М) в координатах приведенного уравнения описываются прямыми линиями. Это подтверждает предположение о двухстадийном механизме метатезиса.
Повышение температуры при метатезисе гексена-1 под действием систем MoO3(1.61 мас.%) / SiO2 С-3 + Al(CH3)3, MoO3 (14 мас.%)/SiO2 ШСК + Al(CH3)3 и MoO3(8.15 мас.%)/Al2O3 + Al(CH3)3 сопровождается повышением активности катализаторов и их эффективности, а также увеличением предельной конверсии гексена-1, но практически не влияет на селективность катализаторов. Зависимости максимальной скорости метатезиса от температуры (в интервале 20оС ÷ 70оС) в координатах уравнения Аррениуса описываются прямыми линиями, что позволило оценить энергию активации процесса Енабл. = 9.4 (MoO3/SiO2 С-3), 9.1 (MoO3/SiO2 ШСК) и Енабл.= 5.5 (MoO3/Al2O3) ккал/моль.
Замечено, что процесс метатезиса альфа-олефинов сопровождается побочными реакциями изомеризации альфа- и «внутренних» олефинов, сометатезиса изомеризованных олефинов с исходными альфа-олефинами и между собой, дейтероводородным обменом, а также алкилированием ароматических углеводородов. Хроматографическим методом установлено, что под действием системы MoO3/SiO2 – Al(C2H5)3 индивидуальный децен-5 превращается в смесь низших и высших гомологов внутренних олефинов (рис. 6).
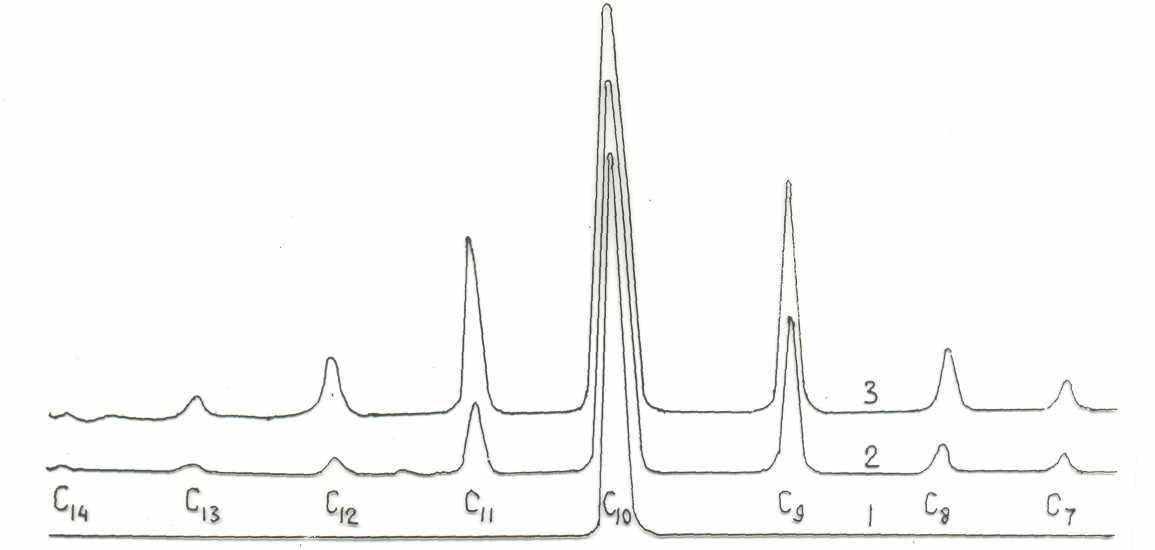
С4- СН=СН-С4 С3-СН=СН-С5 C2-CH=CH-С6
Рис. 6. Хроматограммы децена-5 (1) и продуктов его превращения (2, 3) под действием системы МоО3 (1-63 Mac.%)/SiO2 C-3 при 75 °С в массе. Длительность реакции, мин: 1 - 0; 2 - 30; 3 - 45. MoO3/SiO2 - 3.5 г; Мо - 1.08 мас.% = 0.394 ммоль; активатор - А1(С2Н5)3 - 1.17 ммоль; А1/Мо - 3.0; децен-5 - 0.16 ммоль (1) и 0.08 ммоль (2); Ж.Ф. - 30 мл.
Левая и правая ветвь числового распределения продуктов превращения децена-5 под действием катализатора MoO3/SiO2 – Al(C2H5)3 спрямляются в полулогарифмических координатах. Это свидетельствует о термодинамически-равновесном характере сопряженных процессов метатезиса и изомеризации олефинов. На основании совокупности полученных результатов сделан вывод о бифункциональной металлкарбенгидридной природе активных центров этих процессов: R-CH2=Mo-H. Под действием металлкарбеновой части активного центра протекает каталитический цепной по природе метатезис олефинов, а под действием металлгидридной части активного центра протекают каталитические процессы изомеризации олефинов, дейтеро-водородного обмена и алкилирования ароматических углеводородов с многократной реактивацией исходных частей активных центров.
Среднее число каталитических актов, в которых принимает участие каждая молекула катализатора (эффективность) достигает почти 1000 моль альфа-олефина в расчете на моль МоО3 в катализаторе МоО3 /SiO2 C-3 – Al(CH3)3. Эту характеристику катализатора можно повысить путем диспропорционирования на одном и том же катализаторе нескольких порций альфа-олефина, а также путем многократной окислительной реактивации катализатора. Суммарная эффективность катализатора MoO3 (1.63 мас.%)/SiO2 КСМ (r < 0.25 мм) – Al(CH3)3 после семи реактиваций достигает 2000 моль альфа-олефинов в расчете на 1 моль МоО3 в катализаторе. Приведенная величина эффективности является, видимо, нижним ее пределом и может быть увеличена за счет повышения числа реактиваций и температуры осуществления процесса метатезиса. В настоящее время достигнуты следующие расходные показатели: расход молибдена составляет 0.17 кг на одну тонну переработанного гексена-1; триметил- или триэтилалюминия - 3.60-4.10 кг на одну тонну переработанного гексена-1.
Стадийный механизм метатезиса гексена-1 в децен-5 под действием системы MoO3/SiO2 + Al(C2H5)3. Литературные данные и выполненные нами исследования свидетельствуют о том, что активные центры метатезиса альфа-олефинов имеют металлкарбенгидридную природу и, что цепной по природе процесс метатезиса включает следующие стадии:
- Алкилирование МоО3 триэтилалюминием:
 CH2-CH3
CH2-CH3  Mo = O + Al(C2H5)3 ® Mo
Mo = O + Al(C2H5)3 ® MoO- Al(C2H5)2
- Образование карбенгидридных активных центров метатезиса:
CH2-CH3 H Mo ® CH3 – CH = MoO- Al(C2H5)2 O Al(C2H5)2
- Координация гексена-1 на активном центре метатезиса
 СН3 – СH = Mo CH3 – CH = Mo
СН3 – СH = Mo CH3 – CH = Mo®
C4H9 – CH = CH2 C4H9 – CH = CH2
- Инициирование метатезиса

CH3 – CH = Mo Mo СH3 - CH
® ║ + ║
C4H9 – CH = CH2 CH2 C4H9 - CH
- Повторная координация гексена-1 на активном центре метатезиса
и выделение этилена:
Mo CH – C4H9 Mo = CH - C4H9
║ + ║ ®
CH2 CH2 CH2 = CH2
- Метатезис гексена-1 в децен-5
Mo = CH – C4H9 Mo CH – C4H9
® ║ + ║
СH2 = CH – C4H9 CH2 CH – C4H9
Гексен-1 Децен-5