
Нарушения обмена триптофана (Трп)
| Вид материала | Документы |
- Нарушения липидного обмена выявляются у людей с самыми различными заболеваниями. Эти, 238.5kb.
- Тема лекций Лектор 1 Введение в патологию. Предмет и методы патологии. Болезнь и здоровье., 78.3kb.
- Термодинамика протолитических и координационных равновесий l-аланина, D,L -триптофана,, 330.9kb.
- Ьной недостаточностью инсулина и характеризующееся грубым нарушением обмена углеводов, 81.52kb.
- Тематический план лекций по патологической анатомии для студентов факультета иностранных, 15.46kb.
- Нарушения обмена биометаллов при вибрационных воздействиях и их коррекция 03. 00., 236.45kb.
- Сахарный диабет и беременность, 134.09kb.
- Роль нарушения обмена жирных кислот в развитии дислипопротеинемий, 283.9kb.
- Рефератов, историй болезни, литературы, обучающих программ, тестов, 140.38kb.
- При нарушении функций печени и желчного пузыря, 82.35kb.
Нарушения обмена триптофана (Трп).
Трп – незаменимая АМК имеет 2 пути метаболических превращений – серотониновый (до образования серотонина) и кинурениновый (с образованием как промежуточного продукта – кинуренина) (рис. 3). Второй путь приводит к синтезу НАД (никотинамиддинуклеотида), что уменьшает потребность организма в потреблении витамина РР, из которого, как вы знаете, образуется НАД.
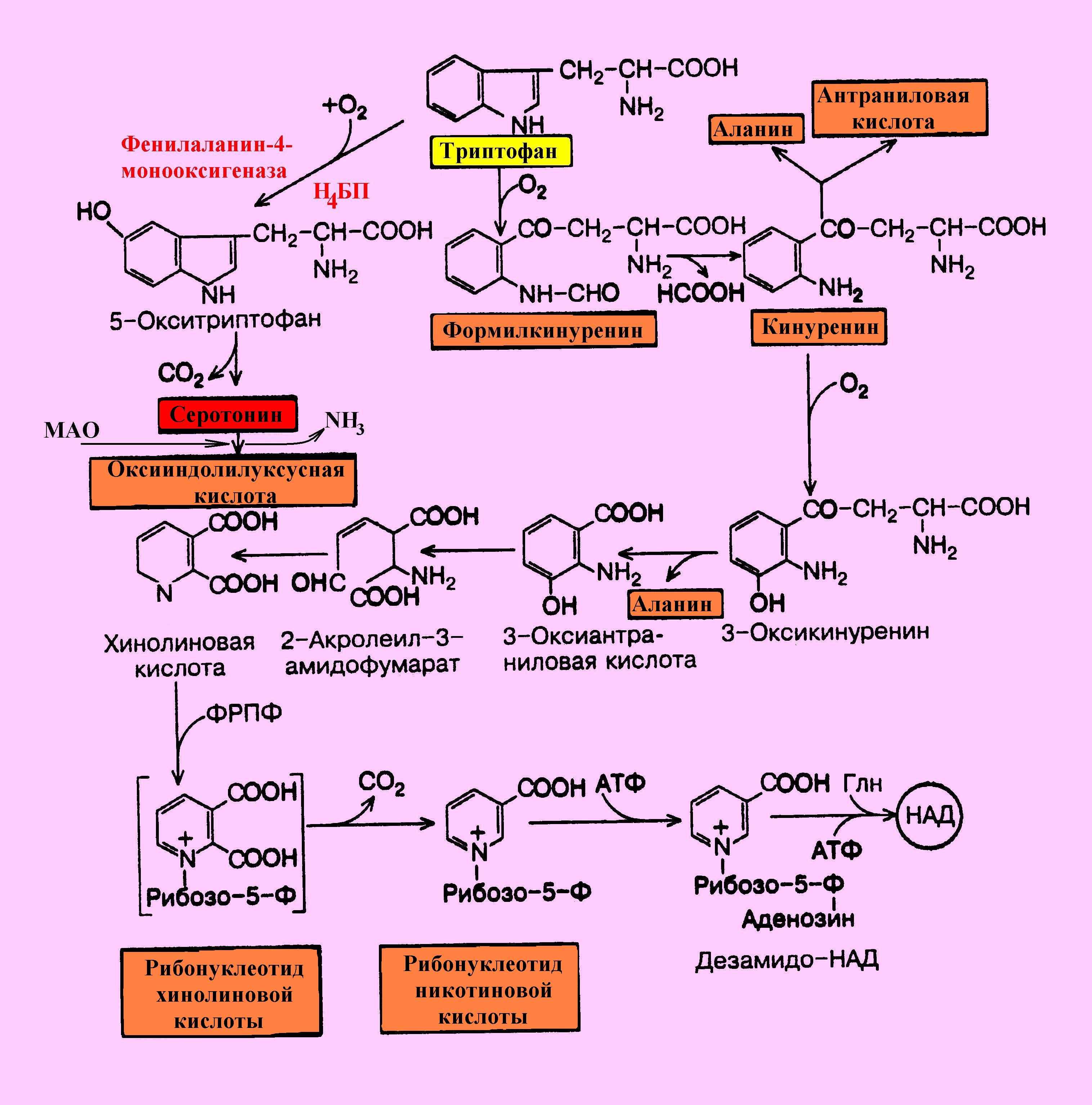
Рис. 3. Пути превращения триптофана.
В «серотониновом» пути метаболизма Трп цепь метаболических превращений открывается фенилаланин-4-монооксигеназой. Следует отметить, что фенилаланин-4-монооксигеназа, участвующая в метаболизме Фен как мы уже рассмотрели, обладает специфичностью и по отношению к Трп, образуя 5-окситриптофан, (см. схему выше), который затем декарбоксилируется до серотонина. Поэтому дефект гена фенилаланин-4-монооксигеназы также как и наследственные нарушения синтеза Н4БП приводят не только к развитию ФКУ, но и к нарушениям «серотонинового» пути метаболизма триптофана прежде всего к нарушению синтеза серотонина и затем мелатонина.
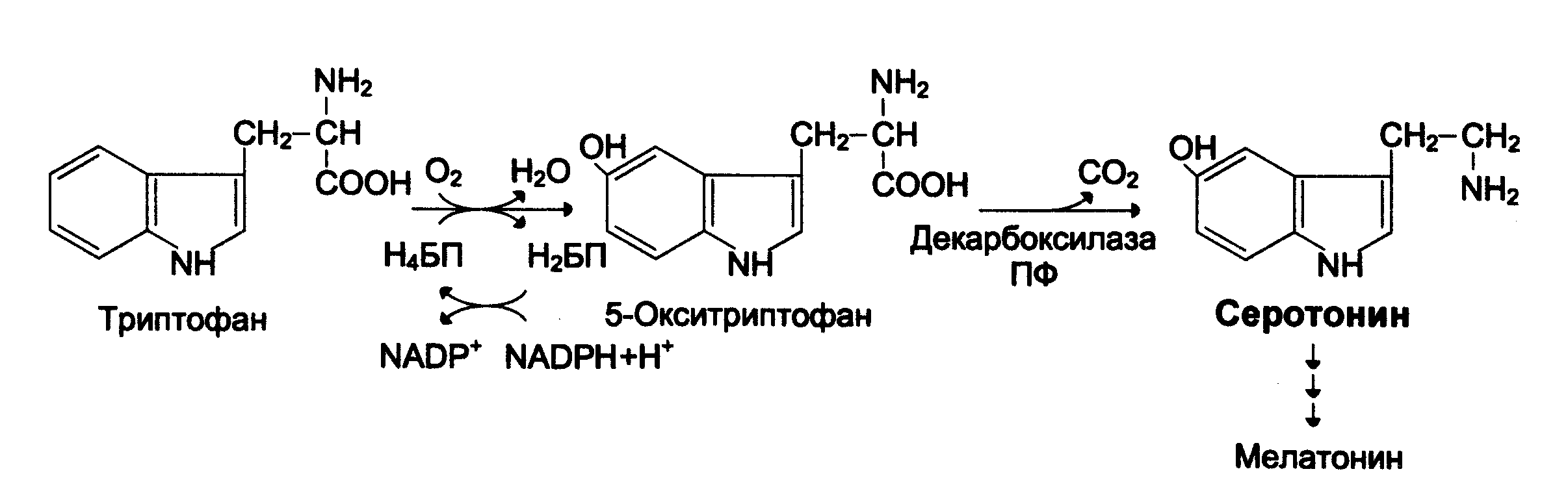
Нарушение синтеза серотонина имеет большое значение для организма так как серотонин стимулирует сокращение гладкой мускулатуры, обладает сосудосуживающим действием, регулирует АД, температуру тела, дыхание, обладает антидепрессантным действием.
Кроме того, серотонин превращается в мелатонин – гормон эпифиза, регулирующий суточные и сезонные изменения метаболизма организма и участвующий в регуляции репродуктивной функции. Мелатонин обладает антиоксидантным действием и может ингибировать процессы ПОЛ.
В норме по серотониновому пути окисляется ~ 1% Трп. Но важность его очень велика и нарушения этого пути представляют большую опасность для организма. Так, при злокачественных новообразованиях ~ 60% Трп окисляется по серотониновому пути. При этом наблюдается снижение образования из Трп НАД, симптомы пеллагры, а также отрицательный азотистый баланс.
Однако 95% Трп в норме окисляется по кинурениновому пути, который начинается с окисления Трп под действием гемсодержащего фермента триптофан-2,3-диоксигеназы с образованием формилкинуренина, распадающегося при участии формамидазы на муравьиную кислоту и кинуренин, окисляемый далее в 3-оксикинуренин. Дальнейшие превращения 3-оксикинуренина связаны с пиридоксалевым ферментом кинурениназой, гидролизующей его на аланин и 3-оксиантраниловую кислоту, которая через ряд промежуточных продуктов превращается в хинолиновую кислоту, т.е. в непосредственный предшественник рибонуклеотида никотиновой кислоты с последующим образованием НАД. Недостаточность витамина В6 приводит к утрате способности к катаболизму 3-оксикинуренина. В этом случае идет образование в норме отсутствующего метаболита – ксантурената, уровень которого в моче млекопитающих и человека появляется при недостаточном содержании в пище витамина В6.
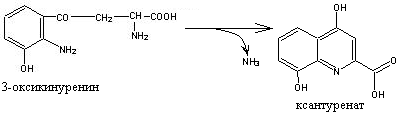
Ксантуренат, может оказывать токсическое действие на -клетки панкреатических островков, являясь одним из патогенетических факторов развития диабета.
Болезнь, связанная с наследственным нарушением транспорта триптофана носит название болезни Хартнупа. Характерно появление сыпи на коже, мозжечковой атаксии и умственной отсталости. Моче больных содержит значительно повышенные количества индолилацетата, индолилацетилглутамина и триптофана. Возможна терапия высокими дозами ниацина (витамин РР).
Нарушения обмена АМК с разветвленной цепью
«Болезнь кленового сиропа» - связана с нарушение обмена АМК с разветвленной цепью: Лей, Иле и Вал. Обмен АМК с разветвленной цепью осуществляется не в печени, как для большинства АМК, а в мышечной, жировой тканях, почках и ткани мозга и включает трансаминирование под действием специфической аминотрансферазы АМК с разветвленной цепью с образованием соответствующих -кетокислот, которые декарбоксилируются с образованием ацил-КоА-производных жирных кислот под действием высокоспецифичного дегидрогеназного комплекса.
«Болезнь кленового сиропа» связана с нарушением декарбоксилирования вследствие синтеза дефектного дегидрогеназного комплекса -кетокислот с разветвленной цепью, что приводит к увеличению накопления в крови Лей, Иле, Вал, -кетокислот и к их экскреции с мочой, издающей запах кленового сиропа. Болезнь встречается редко, обычно – в раннем детском возрасте, приводит к нарушению функции мозга и летальному исходу, если не ограничивать или полностью не исключить поступление с пищей Лей, Вал, Иле.
Наследственные дефекты всасывания АМК в почках.
Хорошо известны цистиноз и цистинурия.
Цистиноз (синдром Абдергальдена-Фанкони).
Основной метаболический дефект связан с врожденным нарушением реабсорбции почти всех аминокислот (за исключением циклических) в канальцах почек, следствием этого являются повышение содержания в моче почти всех АМК – аминоацидурия (при этом экскреция АМК возрастает в 5-10 раз, тогда как цистина и цистеина в 20-30 раз). Наблюдается и избирательное отложение цистина во многих тканях и органах.
Согласно полученным в последнее время данным, причиной болезни является нарушение функции лизосом.
Цистинурия (цистин-лизинурия) – наследственное заболевание. Экскреция с мочой цистина, лизина, аргинина и орнитина в 50 раз выше нормы. Считается, что заболевание связано с нарушением обратного всасывания цистина, лизина, аргинина и орнитина, который, возможно, происходит в общем для них «участке» реабсорбции, поэтому вместо термина цистинурия в настоящее время предпочитают термин цистин-лизинурия. В ходе заболевания у больных могут образовываться цистиновые камни в почечных канальцах.
5-Оксипролинурия определяется дефицитом гена 5-оксопролиназы, катализирующей реакцию превращения 5-оксопролина в глутаминовую кислоту, в -глутамильном цикле, включающий синтез глутатиона (-глутамилцистеинилглицина - GSH) из глутаминовой кислоты, цистеина и глицина при участии ферментов -глутамилцистеинсинтетазы и глутатионсинтетазы.
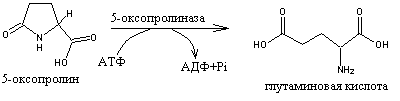
При этом 5-оксопролин образуется в результате того, что -глутамилтранспептидаза (-глутамилтрансфераза), локализованная в цитоплазматической мембране (почечных канальцах, слизистой оболочке кишечника и др. тканей) переносит -глутамильную группу от GSH или другого -глутамильного пептида на транспортируемую АМК.
Комплекс -глутамил-АМК после переноса через биомембрану распадается внутри клетки под действием -глутамилциклотрансферазы на свободную АМК и 5-оксопролин. Благодаря возможности ресинтеза GSH, требующего затраты АТФ, цикл может повторяться многократно, транспортируя значительные количества АМК. Дефицит 5-оксопролиназы делает это невозможным или малоэффектным.
Заболевание наследуется по аутосомально-рецессивному типу. В норме недетектируемый в моче 5-оксопролин в случае заболевания выводится в концентрации до 50 ммоль/сут. Клинические проявления: хронический метаболический ацидоз, гемолитическая анемия, неврологические симптомы. Повышение роста экскреции 5-оксопролина с мочой наблюдается также в случае дефицита гена глутатионсинтетазы, может сопутствовать синдрому гомоцистинурии.