
«применение информационных технологий в биохимических исследованиях»
| Вид материала | Реферат |
- «Применение информационных технологий в социологических исследованиях», 304.23kb.
- Применение информационных технологий в исторических исследованиях, 413.67kb.
- «Применение информационных технологий в религиоведческих исследованиях», 277.44kb.
- «Применение информационных технологий в преподавании литературы», 318.23kb.
- М. В. Моу аннинская сош №6. Всовременное время всё чаще поднимается вопрос о применении, 33.29kb.
- Муниципальная целевая программа «Развитие и применение информационных технологий, 1251.26kb.
- Темы фамилия применение геоинформационных технологий в создании муниципальных информационных, 37.02kb.
- «применение информационных технологий в банковской системе», 242.84kb.
- «применение информационных систем и технологий впроцессе подготовки отчетности по мсфо», 474.4kb.
- «Применение современных информационных технологий при изучении вопросов нетарифного, 316.46kb.
Входная область анализирует фрагмент последовательности размером 15 букв. Предсказание относится к центральному остатку (наверху, отмечен стрелкой). Затем окно сдвигается на одну позицию вправо по последовательности и делается следующее предсказание. Каждой из 15 позиций в окне соответствует 20 нейронов, один из которых активен.
Скрытая область содержит ≈100 нейронов, соединенных с вводом и выводом. Каждый нейрон в скрытой области соединен с каждым нейроном областей ввода и вывода (показаны не все связи).
Область вывода состоит только из трех нейронов, которые просто фиксируют предсказание — спираль, лист, или ни то ни другое.
Важной информацией, которая может быть использована при предсказании вторичной структуры, является эволюционная информация. «Множественное выравнивание» содержит в себе гораздо больше информации, чем одна последовательность. Сохранение вторичной структуры в родственных белках означает наличие связи последовательность — структура, и это позволяет делать более строгие предсказания. Большинство методов предсказания вторичных структур, основанных на нейронных сетях, имеют на входном слое не только информацию о степени консервативности позиции, но и профильные веса [1].
Показано также, что использование двух тандемных нейронных сетей позволяет учитывать корреляцию конформаций соседних остатков. Предсказания состояний нескольких последовательно идущих остатков с помощью сети, аналогичной показанной на рисунке 3, комбинируются с помощью еще одной сети, которая формирует окончательный результат. Тест на зрелость методов предсказания может быть проведен полностью автоматически.
Некоторые вычислительные методы продуцируют только предварительное грубое предсказание структуры и требуют вмешательства человека для формирования окончательного результата. Другие методы полностью автоматические.
Есть множество Web-ресурсов, которые принимают последовательность и возвращают предсказание структур и функций белков. PredictProtein — одна из таких систем, использовавшихся для предсказания вторичной структуры белка MutS. Пользователи предоставляют первичные последовательности белков, их субъединиц или даже участков на данный сервер, на основе чего PredictProtein с использованием большого пакета различных программ производит полный анализ первичной последовательности данного белка, позволяет установить в её пределах функциональные мотивы (PROSITE), регионы низкой и высокой сложности (SEG), сигналы ядерной локализации, регионы, лишенные регулярной структуры (NORS), а также предоставляет возможную вторичную структуру, даёт информацию о доступности растворителя к различных областям белковой молекулы, о глобулярных областях, трансмембранных спиралях, биспиральных (coiled-coil) регионах, структурных switch областях, дисульфидных связях, субклеточной локализации и возможных функциях [7].
По запросу также дополнительно можно осуществить распознавание фолда методом «трединга», установить предназначение CHOP доменов в данном конкретном случае, предсказать контакты между трансмембранными участками и между остатками [7].
Ниже приведён список программ, которые используются на ресурсе PredictProtein, и их краткое описание:
- PROSITE – база данных функциональных мотивов;
- ScanPROSITE обнаруживает все функциональные мотивы в задаваемой последовательности, которые аннотированы в базе данных ProSite db;
- SEG подразделяет задаваемые последовательности на области высокой и низкой сложности;
- ProDom является базой данной предполагаемых белковых доменов; при помощи BLAST осуществляется поиск в пределах этой базы данных тех доменов, которые соответствуют белку исследователя;
- MaxHom – это программа динамических «множественных выровненных» последовательностей; она позволяет найти в имеющейся базе данных последовательности, которые соответствуют последовательностям такого же характера анализируемого белка;
- MView позволяет превратить «множественные выровненные» последовательности в «разукрашенные» выходные данные в формате HTML;
- PHD подходящая программа для прогнозирования 1D структуры (вторичной структуры, доступности растворителю) на основе «множественных выровненных» последовательностей
- PHDsec прогнозирует вторичной структуру на основе «множественных выровненных» последовательностей
- PHDacc подходящая программа для прогнозирования доступности каждого аминокислотного остатка по отношению к растворителю на основе «множественных выровненных» последовательностей;
- PHDhtm предсказывает положение и топологию трансмембранных спиралей на основе «множественных выровненных» последовательностей.
- PROF - подходящая программа для предсказания 1D структуры (вторичной структуры, доступности растворителю) на основе «множественных выровненных» последовательностей.
- PROFsec прогнозирует вторичной структуру на основе «множественных выровненных» последовательностей.
- PROFACC предсказывает доступность каждого аминокислотного остатка по отношению к растворителю на основе «множественных выровненных» последовательностей.
- GLOBE осуществляет предсказание глобулярности белка
- DISULFIND предсказывает наличие в белковой структуре дисульфидных мостиков, основываясь на двухэтапный процесс
- ASP обнаруживает области, в которых наиболее вероятно осуществляются конформационные переходы [7];
Таким образом, алгоритм действий при моделировании структуры белков в случае использования PROF ресурса следующий [7]:
- Необходимо предоставить запрос на сайт www.predictprotein.org в виде аминокислотной последовательности, которую можно получить в любой базе данных, например, PDB.
- Далее результаты выполненной по запросу работы отправляются либо по электронной почте пользователю, или предоставляются в интерактивном режиме непосредственно на сайте.
2.2 Моделирование по гомологии
Построение модели по гомологии — полезный метод для предсказания структуры белка по известной последовательности, когда исследуемый белок состоит в родстве хотя бы с одним белком, для которого известны и последовательность, и структура. Если белки являются близкими родственниками, то известные белковые структуры (называемые родительскими) могут служить основой для модели исследуемого белка. И хотя качество модели зависит от степени сходства последовательностей, оценить это качество возможно до экспериментальной проверки. Поэтому знание того, какое качество модели требуется приложением, для которого она предназначена, позволяет с высокой долей вероятности предсказать успешность выполнения задачи [1].
Шаги моделирования по гомологии следующие:
- «Выровнять» аминокислотные последовательности исследуемого белка и белка (белков) с известной структурой. В большинстве случаев вставки и делеции будут наблюдаться в петлях между α-спиралями и β-тяжами.
- Определить сегменты основной цепи, содержащие вставки или делеции. Сшивание этих участков с основной цепью известного белка (матрицы) создает модель основной цепи исследуемого белка.
- Заменить боковые цепи мутировавших остатков. Для немутировавших остатков сохранить конформацию боковых цепей. Мутировавшие остатки склонны сохранять конформацию боковой цепи, и это можно использовать при моделировании. При этом сейчас также доступны вычислительные методы поиска подходящей конформации боковой цепи среди возможных комбинаций.
- Проверить модель (и визуально, и с помощью программ), чтобы выявить значительные наложения атомов. Насколько возможно, устранить подобные наложения вручную.
- Уточнить модель методом ограниченной минимизации энергии. Роль этого шага — установить точное геометрическое расположение в тех местах, где были соединены участки главной цепи, и позволить боковым цепям слегка перемещаться, чтобы занять удобное положение. На самом деле эффект этой процедуры только косметический: минимизация энергии не устранит серьезных ошибок в такой модели [1].
По сути, модель нового белка строится путем внесения минимальных изменений в доступную структуру матрицы. К сожалению, существенно улучшить такую модель непросто. Эмпирическое правило гласит, что если две последовательности идентичны хотя бы на 40-50%, описанная процедура дает модель, достаточно точную для использования во многих приложениях. Если же сходство ниже, то ни описанная процедура, ни какой-либо другой доступный на данный момент алгоритм, не дадут детально точной модели, исходя из доступных структур родственных белков [1].
Структуры большинства белковых семейств содержат как относительно постоянные, так и более вариабельные участки. Ядро структуры семейства сохраняет топологию укладки, хотя и может быть искажено, периферия же может быть целиком сложена заново. Располагая единственной прародительской структурой, можно относительно достоверно моделировать консервативную часть исследуемого белка, но построить модель вариабельной части уже не удастся. Более того, непросто предсказать, какие участки являются вариабельными, а какие — консервативными. Более предпочтительна ситуация, когда несколько родственных белков с известной структурой могут выступать в качестве родителей для моделирования по гомологии. Они выявляют внутри семейства участки с консервативной и вариабельной структурой. Наблюдаемое распределение структурной вариабельности среди родительских структур диктует подходящее распределение ограничений применительно к моделированию [1].
Развитое программное обеспечение для моделирования по гомологии доступно.
SWISS-MODEL — это Web-сайт, который принимает на входе аминокислотную последовательность исследуемого белка, определяет, существует ли подходящая для моделирования по гомологии родительская структура (структуры), и, если существует, выдает на выходе набор координат исследуемого белка. SWISS-MODEL разработали Т. Schwede, М. С. Peitsch и N. Guex в Женевском институте биомедицинских исследований [1, 2, 12].
В зависимости от сложности задачи, которую необходимо решить при моделировании пространственной структуры белка, интересующего исследователя, на сайте SWISS-MODEL может быть использован один из трёх возможных алгоритмов действия: автоматическое моделирование, «способ выравнивания» и «проектный способ». Эти способы отличаются по степени участия пользователя в процессе [2, 12].
- Автоматическое моделирование подходит в тех случаях, когда имеется достаточно высокая степень сходства между исследуемым белком и белком, выступающим в роли матрицы (образца для сравнения). Как правило, если целевой белок и матрица имеют более 50 % идентичности в последовательностях, то автоматические «выравнивания» последовательностей являются достаточно надежными [12].
Для подачи запроса требуется только наличие аминокислотной последовательности целевого белка или кода доступа (если последовательность данного белка хранится в базе UniProt) в качестве вводимых данных. Программный пакет сервера автоматически выберет подходящие матрицы, используя предел величины Е (E-value limit) из Blast, экспериментальные данные, данные о связываемых молекулах субстратов или различные конформационные состояния белка-матрицы [12].
В зависимости от того, как планируется применять получаемую модель, обязательно нужно выбрать белок-матрицу в правильной конформации. Для чёткого определения структуры белка-матрицы пользователь может предоставить id из ExPDB), например, 1akeA, где PDB-ID - 1ake, Chain ID – А [12].
Примером автоматического предсказания с помощью SWISS-MODEL является предсказание структуры трансмембранной протеазы 3 человека (TMPRSS3), представленное на рисунке 4.
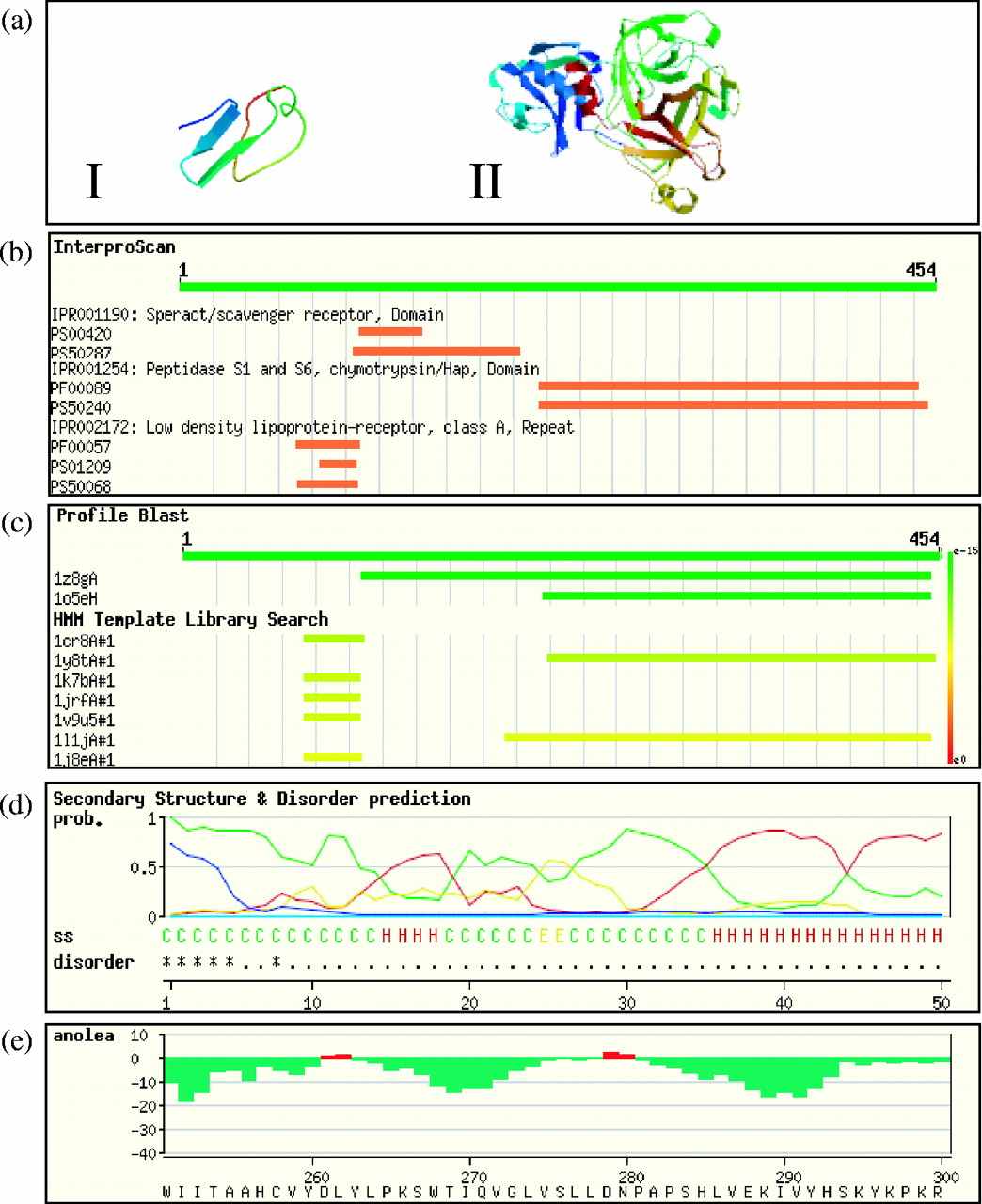
Рисунок 4. Графический результат моделирования белковой структуры, осуществленный в рамках «рабочего места» на SWISS-MODEL.
Типичные этапы эксперимента по моделированию:
(a) Представление в «ленточной» форме трёх доменов, смоделированных для целевого белка – трансмембранной протеазы 3 человека (TMPRSS3): I, LDL рецепторный домен и II, рецептор – связывающий домен в комплексе с протеазным доменом.
(b) программа IprScan последовательности целевого белка позволила обнаружить три домена в целевом белке.
(c) поиск, основанный на аминокислотной последовательности, в библиотеке матриц показал наличие двух сегментов с подходящими матричными структурами
(d) Представлены вторичная структура и предсказание неупорядоченности целевого белка
(e) на графике представлена величина «anolea», позволяющая оценить качество окончательной модели.
- «Способ выравнивания» позволяет пользователю проверить несколько альтернативных выравниваний и оценить качество полученных моделей для достижения оптимального результата. «Способ множественных выравниваний» последовательности является общим средством во многих молекулярно-биологических исследованиях. Если трехмерная структура известна, по крайней мере, хотя бы для одного из родственных белков, то это выравнивание может быть использовано в качестве исходной точки для сравнительного моделирования на основе «способа выравнивания» [12].
Для облегчения использования «способа выравнивания» в различных форматах, подача данных осуществляется как трехэтапный процесс:
- Подготовка множественного выравнивания последовательности:
- Запрос должен содержать, по крайней мере, целевую последовательность и последовательность белка-матрицы
- Нужно использовать любое из возможных средств выравнивания, например, T_COFFEE (Cedric Notredame)
- Убедиться в том, что у последовательностей "разумное" названия
- Необходимо предоставить «выравнивание» на «рабочее место», где также указывается использованный «способ выравнивания».
- Возможные форматы: FASTA, MSF, CLUSTALW (рисунок 5), PFAM and SELEX
- Можно либо загрузить файлы, либо скопировать и вставить последовательности
- Обязательно нужно правильно указать формат выравнивания,

Рисунок 5. «Выровненная» последовательность белка с использованием программы CLUSTALW.
- Выбрать целевой белок и белок-матрицу
- Выравнивание (в той форме, какой оно было интерпретировано сервером) должно отражаться в нижней части страницы
- Сценарий программы самостоятельно попытается определить правильные названия, основываясь на данных, предоставленных пользователем;
- Выбрать название той последовательности, которая является целевой (target template)
- Выбрать название последовательности, которая будет являться матрицей (template sequence, например, 1crnA), при этом совсем не обязательно в качестве названий использовать ID из PDB, можно использовать названия, которые нравятся пользователю.
- Необходимо определить структуру матрицы, которой принадлежит эта последовательность. Эта матрица обязательно должна быть частью библиотеки матриц ExPDB, поэтому необходимо воспользоваться библиотекой матриц SWISS-MODEL
- Также необходимо указать правильное название ID цепи белка-матрицы, необходимо отметить, что названия цепей должны быть указаны заглавными буквами.
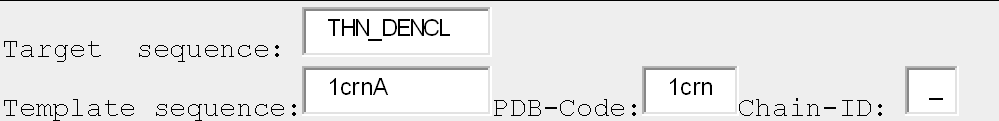
Рисунок 6. Правильно заполненная форма запроса.
Сверху указано название целевой белковой последовательности, внизу последовательности-матрицы, далее располагается PDB ID белка-матрицы (1 crn в данном случае), в приведенном примере не указывается цепь белка-матрицы для анализа
- Проверка выравнивания и подтверждение запроса
- «Выравнивание» внизу страницы должно представлять правильное отображение структуры матрицы по отношению к целевой последовательности. Это необходимо тщательно проверить перед подтверждение запроса.
- Обычно необходимо предоставить адрес электронной почты для оформления заявки на SWISS-MODEL.
Сервер построит модель исключительно на основе «выравнивания» [12].
- «Проектный способ»: используется в тех случаях, когда правильное «выравнивание» между целевым белком и матрицей не может быть определено на основе метода последовательностей, визуальная проверка и ручная обработка «выравнивания» может существенно помочь улучшить качество модели, получаемой на выходе. Файлы «проектного способа» содержат матричные структуры и «выравнивание» между целевым белком и матрицей. Данные файлы формируются при помощи программы DeepView (Swiss-PdbViewer) или средств выбора матриц, которые имеются в рабочей области. Файлы «проектного способа» предоставляются на выходе в формате, заданным по умолчанию моделирующим сервером, что позволяет анализировать и каждый раз улучшать модели, сгенерированные при помощи «автоматического моделирования» и «способа выравнивания» [12].
2.3 Распознавание фолда
Поиск последовательности в базе данных последовательностей и поиск структуры в базе данных структур — это задачи, имеющие решения. Смешанные задачи (поиск по структуре в банке последовательностей или по последовательности в банке структур) менее очевидны. Они требуют метода для оценки совместимости данной последовательности с данным способом фолда [1].
Цель состоит в выделении существенного набора последовательностей и структур. Ожидается, что белки, имеющие один и тот же паттерн, имеют схожие структуры.
J. U. Bowie, R. Lüthy и D. Eisenberg проанализировали окружение каждой позиции в известных белковых структурах и соотнесли с набором предпочтений двадцати аминокислот в структурном контексте [1].
Имея белковую структуру, можно классифицировать окружение каждой аминокислоты по трем отдельным категориям:
водородные связи основной цепи, т. е. вторичная структура;
- степень погруженности внутрь или экспонированности на поверхность белковой глобулы;
- полярная/неполярная природа окружения [1].
Возможны три варианта вторичной структуры: α-спиралъ (helix), β -слой (sheet) и иное. Авторы определяют 6 классов аминокислот на основе доступности и полярности окружения. Боковые цепи каждого из этих шести классов могут быть в любом из трех типов вторичной структуры. Таким образом, всего получается 18 классов. Если отнести каждую боковую цепь к одному из 18 классов, то можно, пользуясь алфавитом из 18 букв, создать описание белковой структуры, называемое профилем трехмерной структуры (ЗD-профилем). К «последовательностям», в которых таким образом закодированы структуры, можно применить алгоритмы, разработанные для поиска последовательностей. Например, можно попытаться выровнять две далекие друг от друга родственные последовательности путем выравнивания их ЗD-профилей, а не самих аминокислотных последовательностей. Метод ЗD-профилей превращает белковые структуры в одномерные объекты, не сохраняющие точно ни последовательность, ни структуру молекул, из которых они были получены [1].
Далее необходимо соотнести ЗD-профиль с набором известных последовательностей и структур. Очевидно, что некоторые аминокислоты «не рады» находиться в определенных местах; например, заряженная боковая цепь не может быть спрятана внутри совсем неполярного окружения. Остальные предпочтения не столь четки, поэтому необходимо составить таблицу предпочтений на основе статистического обзора библиотеки, содержащей белковые структуры высокого качества [1].
Когда у исследователя есть последовательность и он хочет оценить вероятность того, что она принимает, скажем, фолд глобина, то из ЗD-про-филя известной структуры миоглобина кашалота он знает класс окружения у каждой позиции в последовательности. Далее исследователь рассматривает частичное «выравнивание» неизвестной последовательности с миоглобином кашалота и предполагает, что первому остатку миоглобина соответствует в неизвестной последовательности остаток фенилаланина. В ЗD-профиле класс окружения первого остатка миоглобина следующий: экспонированная боковая цепь, нет вторичной структуры. Можно оценить вероятность нахождения фенилаланина в этом классе окружения, используя таблицу предпочтений отдельных аминокислот для этого класса ЗD-профилей Распространение этих подсчетов на все позиции и на все возможные выравнивания (не допускающие разрывов внутри участков, имеющих вторичную структуру) дает число, которое оценивает, насколько хорошо данная неизвестная последовательность подходит профилю миоглобина кашалота [1].
Особое преимущество этого метода состоит в том, что он может быть автоматизирован. Новую последовательность можно сравнивать с каждым ЗD-профилем в библиотеке известных фолдов по сути таким же способом, какой отработан для сравнения новой последовательности с библиотекой известных последовательностей [1].
ЗD-профиль, полученный из структуры, весьма опосредованно зависит от аминокислотной последовательности, но вместе с тем эффективно используется для определения качества структур. Есть два интересных наблюдения:
- Белковые структуры, хорошо соответствующие собственным профилям на родственных белках, дают высокий вес сопоставления самих структур этих белков между собой. Профиль является абстрактным свойством семейства, а не только индивидуально белка.
- Когда родственная последовательность плохо соответствует профилю, полученному из экспериментальной структуры этой последовательности, то, по-видимому, в структуре есть ошибка. Позиции, где профиль не соответствует последовательности, могут указывать на область, где находится ошибка [1].
Методом распознавания фолда является трединг (threading — протягивание). Основная идея данного метода состоит в том, чтобы построить много грубых моделей для данной последовательности, используя всевозможные выравнивания с последовательностями, для которых известна структура. Систематическое исследование множества возможных выравниваний определило название метода. Можно представить, что осуществляется аккуратное протягивание белковой последовательности через известную трехмерную структуру. При этом допустимы вставки и делеции, но если протягивание достаточно мягкое, то метафора «протягивания» остается в силе [1].
Как трединг, так и моделирование по гомологии, имеют дело с трехмерными структурами, индуцированными выравниваниями искомой последовательности и последовательности, для которой трехмерная структура определена. Моделирование по гомологии концентрируется на множестве выравниваний и имеет целью построение детальной структуры. Трединг использует множество различных выравниваний и работает только с грубыми моделями, иногда даже не построенными явно. Можно привести краткое сравнение данных подходов [1].
| Моделирование по гомологии | Трединг |
| Сначала найти гомологов | Проверить всех возможных партнеров |
| Построить оптимальное выравнивание | Проверить все возможные выравнивания |
| Оптимизировать одну модель | Оценить много грубых моделей |
Для успешного распознавания с использованием трединга требуется:
Метод для оценки моделей, позволяющий выбрать одну.
- Метод калибровки весов, чтобы можно было понять, на сколько выбранная модель хороша [1].
Было испытано несколько аппроксимаций взвешивания. Одна из наиболее эффективных основана на эмпирической оценке близости аминокислотных остатков, полученной из анализа известных структур. Наблюдения над межостаточными расстояниями в известных структурах для всех 20 х 20 пар типов остатков. Для каждой пары остатков было построено распределение вероятностей пространственных расстояний между остатками. Например, для пары Leu-Ile были рассмотрены все Leu и Ile во всех структурах и были вычислены пространственные расстояния между Сβ-атомами и расстояния по последовательности. Коллекция этих данных позволила построить оценку, насколько хорошо расстояния в модели соответствуют расстояниям в известных структурах [1].
Распределение Больцмана связывает энергию и вероятность. При трединге — из вероятности выводится энергия. Эта энергетическая функция используется для оценки качества модели [1].
Для каждой структуры из библиотеки процедура находит соответствие остатков, доставляющее минимум энергии. Хотя это и является задачей выравнивания, нелокальность взаимодействий не позволяет применить здесь метод динамического программирования [1].
Наилучшие методы предсказания фолда единообразно эффективны. Они включают методы, основанные на трединге, но не ограничиваются им.
Программа ROSETTA — продукт лаборатории D. Baker для предсказания структуры белка по аминокислотной последовательности. Данная программа использует информацию об уже расшифрованных структурах. В настоящий момент, данный программный продукт опережает свои аналоги на несколько корпусов [1, 3].
ROSETTA, используя данные об уже имеющихся структурах, сначала предсказывает структуру отдельных фрагментов, объединяя их впоследствии в единую структуру. Вначале последовательность разбивается на фрагменты от 3 до 9 аминокислот и происходит поиск схожих фрагментов в белках с известной структурой. Поскольку фрагменты достаточно короткие, то никаких предположений о родственных связях между белками не делается. Исходя из возможных вариантов структуры отдельных фрагментов, рассчитываются возможные варианты структуры белка в целом [1,3].
ROSETTA использует для анализа таких комбинаций метод Монте-Карло. Основная идея заключается в том, что структура, полученная наибольшее количество раз в ходе независимых испытаний Монте-Карло, и будет наиболее правдоподобной моделью [1].
Сейчас полностью автоматизированное выполнение программы Rosetta для предсказания структуры белков осуществляется с помощью Internet сервиса Robetta, который также включает средства анализа, позволяющие сделать выводы о структуре белков на основе геномных данных. Программа Rosetta была эффективно использована для описания структур CASP-5 [3].
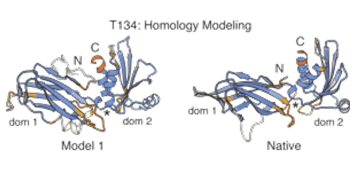
Рисунок 7. Результаты построения пространственных структур для белков CASP-5 с помощью программы ROSETTA
Слева изображена модель пространственной структуры белка, построенная программой ROSETTA, а справа – нативная пространственная структура данного белка. Таким образом, модель оказалась очень близка нативной пространственной структуре белка.
Сервис Robetta разбивает введенные аминокислотные последовательности белков на отдельные домены и осуществляет построение моделей не только для доменов, последовательности которых гомологичны таковым для белков с известной структурой (сравнительное моделирование – программа Rosetta), но и для доменов, лишенных такой гомологии (метод Rosetta de novo). Кроме того, сервер может определять структуры белков с помощью программы RosettaNMR на основе данных ЯМР, предоставленных пользователем. В качестве результата пользователь получает предсказания для доменов и молекулярные координаты моделей распределения для всей последовательности, которая вводилась в запросе [4, 6, 11].
Алгоритм действий для получения пространственной структуры белка с помощью программы Rosetta на сервисе Robetta следующий [11]:
- Пользователь должен зарегистрироваться на сайте ссылка скрыта прежде, чем сделать запрос на сервис Robetta.
- Пользователь предоставляет аминокислотную последовательность белка, для которого хочет получить пространственную структуру.
Последовательности, предоставляемые на сервер для предсказания структуры, должны быть записаны в формате однобуквенных аминокислот. Они могут быть либо вставлены в форму запроса, либо загружены из файла. Пользователи могут или предоставить последовательность для идентификации отдельных доменов, или для предсказания полной последовательности. У пользователя также имеется возможность указать PDB ID того белка и той цепи, которые будут использоваться для сравнительного моделирования.
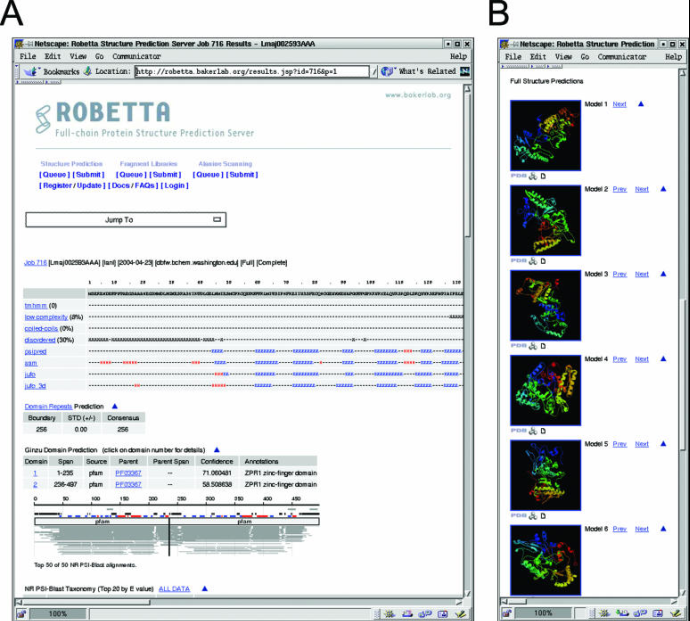
Рисунок 8. Результаты, полученные при использовании сервера ROBETTA для предсказания структуры белка.
В верхней части экрана предоставляются цифровые и статические данные ( A), внизу экрана располагаются непосредственно смоделированные трехмерные белковые структуры (в данном случае первые 6 из 10 предсказанных структур (B)).
- Для загрузки результатов по предсказанию структуры целевого белка на адрес электронной почты пользователя по завершению работы высылается особая ссылка. Поскольку библиотеки фрагментов являются большими (мегабайты), то данные по запросу удаляются с сервера через неделю после завершения работы.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Описанные выше программы, которые широко используются для предсказания структур белков, хорошо зарекомендовали себя, однако не являются единственными.
Так, наряду с программами PROF и PSIPRED для предсказания вторичной структуры белков могут быть использованы PROFsec, PHD, PHDsec и целый ряд других программных пакетов. Точно также обстоит дело и с программами, предназначенными для моделирования по гомологии (кроме сайта SWISS-MODEL с его программным обеспечением используется также, например, сервер MODBASE), и с программами, используемыми для моделирования на основе распознавания фолда (не только ROSETTA, но LINUS и др.)
Проблема прогнозирования структуры белков является весьма актуальной не только в области современной биологии, но и в медицине, фармакологии. Поэтому и неудивительно, что идёт непрерывный процесс совершенствования не только уже имеющегося программного обеспечения в данной области, но и создание нового. И конечно, поскольку существует большое количество программ для прогнозирования, возникает правомерный вопрос о сравнении результатов, полученных с их помощью.
Так, существует проект CASP – программа долгосрочного исследования, предназначенная для оценки методов для предсказания белковых структур. В рамках этой программы кристаллографы и ЯМР-спектроскописты публикуют аминокислотные последовательности белков, структуры которых они расшифровали, но сами структуры держат в тайне до тех пор, пока «предсказатели» не будут готовы представить модели этих белков. Каждые два года последовательности публикуются весной, а к осени предсказания должны быть уже готовы. Затем прогнозы и эксперименты сравниваются и в конце года авторы предсказаний собираются на торжественной конференции для обсуждения текущих результатов и оценки успехов [1, 10].
Предсказания в CASP делятся на три основные категории:
- предсказание вторичной структуры;
- сравнительное моделирование (моделирование по гомологии);
- распознавание укладки и моделирование новых фолдов;
Три эксперта-оценщика, по одному на каждую категорию, сравнивают предсказанную и экспериментально расшифрованную структуры и оценивают качество предсказаний. В число докладчиков входят организаторы, эксперты-оценщики и предсказатели, включая тех, кто только отчасти добился успеха и тех, кто разработал интересный новый метод моделирования [1].
Именно в рамках осуществления проекта CASP в 2000 г. была показана высокая эффективность программ PROF для предсказания структуры домена белка репарации MutS из Thermus aquaticus и ROSETTA для предсказания нового фолда в случае N – концевой половины домена 1 белка человека Xrcc4 из системы репарации ДНК [1, 10].
Стоит отметить, что непрерывный полностью автоматический анализ предсказанных структур (включая предсказание вторичных структур, но не только их) осуществляется Web-сервером ЕVА. Это плод сотрудничества групп из Нью-Йорка и Мадрида. Целью проекта EVA является мониторинг прогресса в этой области и выработка рекомендаций пользователям для использования различных серверов предсказания структуры в разных категориях. EVA может рассматриваться как непрерывный CASP, ограниченный методами, которые могут быть проверены автоматически. Вместе с тем EVA имеет доступ к гораздо большему набору данных, чем САSР. Поэтому ее решения менее подвержены статистическим флуктуациям и трудностям выбора задач в САSР [1,13].
ЗАКЛЮЧЕНИЕ
Таким образом, на основании вышеизложенного материала можно однозначно утверждать, что интенсивное проведение современных молекулярно-биологических, биохимических, биотехнологических и ряда других исследований неразрывно связано с внедрением и эффективным использованием информационных технологий и привело к созданию новой научной области – биоинформатики.
На современном этапе решение проблемы, связанной с предсказанием структуры и свойств белков на основании данных об их аминокислотном составе, осуществляется благодаря усилиям учёных из различных стран, создаются и реализуются программы долгосрочных исследований, такие, как например, CASP. Хорошо зарекомендовало себя использование описанных в данной работе подходов для предсказания структур белков, но вместе с тем не исключена возможность разработки и новых подходов.
Необходимо подчеркнуть, что происходит активное использование и усовершенствование уже имеющихся программ, а также активная разработка нового программного обеспечения, необходимого для определения структуры белков по их аминокислотным последовательностям на основе таких.
Наличие мощных Web-узлов, хранящих необходимое программное обеспечение и обширные базы данных, позволяет осуществлять быструю навигацию имеющейся информации, систематизировать данные, выполнять необходимые исследования и объединять учёных по всему миру.
СПИСОК ЛИТЕРАТУРЫ К РЕФЕРАТУ
- Леск А.. Введение в биоинформатику. – М.: БИНОМ. Лаборатория знаний, 2009. – 318 с.
- Arnold, K., Bordoli, L., Kopp, J., Schwede, T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling // Bioinformatics. – 2006. – Vol. 22, № 2. – P. 195–201
- Bradley, P., Chivian, D., Meiler, J., Misura, K.M., Rohl, C.A., Schief, W.R., Wedemeyer, W.J., Schueler-Furman, O., Murphy, P., Schonbrun, J. et al. Rosetta predictions in CASP5: successes, failures, and prospects for complete automation. // Proteins. – 2003. – 53, Suppl. 6. – P. 457–468.
- Chivian, D., Kim, D.E., Malmstrom ,L., Bradley, P., Robertson, T., Murphy, P., Strauss, C.E., Bonneau, R., Rohl, C.A. and Baker, D. Automated prediction of CASP-5 structures using the Robetta server. // Proteins. – 2003. – 53, Suppl. 6. – P. 524–533.
- Introduction to Protein Architecture: The Structural Biology of Proteins. // Oxford University Press, 2001
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. // Nucleic Acids. – 2004. –July 1; 32, Web Server issue: W526–W531.
- Rost, B., Yachdav G. and Liu J. The PredictProtein Server.// Nucleic Acids Research. – 2004. – 32, Web Server issue: W321-W326.
- McGuffin, L.J., Bryson K., Jones, D.T. The PSIPRED protein structure prediction server.// Bioinformatics. - 2000. – 16. – P. 404-405.
- ctprotein.org/
- ncenter.gc.ucdavis.edu/
- ссылка скрыта
- ссылка скрыта
- c.columbia.edu/eva
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ К РЕФЕРАТУ
310-спираль 10
ASP 20
CASP 5, 34, 37, 38, 39, 40
CLUSTALW 26, 27
DISULFIND 20
ExPDB 5, 24, 27
FASTA 26
GLOBE 20
MaxHom 19
MSF 26
MView 20
PDB 5, 21, 24, 27, 28, 35
PFAM 26
PHD 20, 37
PHDacc 20
PHDhtm 20
PHDsec 20, 37
PredictProtein 18, 19, 40, 44
ProDom 19
PROF 15, 20, 37, 38
PROFACC 20
PROFsec 20, 37
PROSITE 19
PSIPRED 15, 16, 37, 40
ScanPROSITE 19
SEG 19
SELEX 26
SWISS-MODEL 23, 24, 25, 27, 28, 37, 40
T_COFFEE 26
α-спираль 9, 10, 11, 13, 21
αβ-баррелы 11
β-лист 10, 13
β-слой 11
β-тяж 21
π-спираль 10
автоматическое моделирование 23, 29
белок-матрица 24, 26, 27, 28
вторичная структура 9, 10, 13, 14, 15, 16, 17, 18, 20, 25, 29, 30, 37, 38
выравнивание 14, 23, 24, 26, 27, 28, 30, 31, 32, 33
домен 12, 15, 19, 25, 34, 35, 38
ЕVА 5, 38
ЗD-профиль 30, 31
конформационное состояние 24
конформация 13, 14, 18, 22, 24
матрица 5, 21, 22, 23, 24, 25, 27, 28
матричная структура 25, 28
множественное выравнивание 18, 26
множественное выравнивание последовательности 26
моделирование по гомологии 14, 21, 23, 32, 37, 38
модульные белки 12
мотив(ы) 9, 19
неупорядоченные фрагменты 10
первичная структура 9, 10
пространственная структура 13, 14, 23, 34, 35
сегмент(ы) 13, 21, 25
супервторичные структуры 12
трединг 19, 31, 32, 33
третичная структура 10, 12, 14
фолд 13, 14, 19, 29, 30, 31, 33, 37, 38
целевая последовательность 26, 28
целевой белок 8, 15, 16, 23, 24, 25, 27, 28, 36
четвертичная структура 11
ИНТЕРНЕТ-РЕСУРСЫ В ПРЕДМЕТНОЙ ОБЛАСТИ
ссылка скрыта
BioMedNet организован Elsevier Science. Это web-сайт для биологов и медицинских работников. Можно получать новости сайта по E-mail. На этом сайте:
- публикуются обзоры, новости, обзоры конференций;
- имеется хорошая подборка аннотированных web-ресурсов;
- имеется список журналов со свободным доступом (часто временно ради рекламы доступны хорошие журналы), возможность подписаться на "содержание журналов";
- возможен поиск фирм производителей конкретной медико-биологической продукции;
- имеются: MEDLINE; Technical Tips (коллекция молекулярно-биологических протоколов);
- имеется база вакансий с возможностью поиска.
ссылка скрыта
Всемирно известная поисковая система Google. Позволяет производить простой поиск по ключевым словам, возможен вариант расширенного поиска по группам (среди книг, музыкальных файлов или видеофайлов, новостей и т.д.), особым признакам (определение, тип файла) и т.д.
stanford.edu
Сайт секции библиотеки Стэнфордского университета предлагает вниманию пользователей огромную базу материалов, доступных к бесплатному скачиванию в полном объеме. Источниками предлагаемых статей являются 975 журналов, читатели имеют возможность доступа к полным текстам почти 1 435 924 статей, которые перед публикацией получили рецензию экспертов. Возможен быстрый поиск и расширенный поиск (по авторам статей, названиям, цитатам, ключевым словам и т.д.).
ссылка скрыта
Практическая молекулярная биология. Сайт является незаменимым для биохимиков, генетиков, микробиологов и молекулярных биологов. Это крупнейшая биологическая база данных. Сайт содержит подробный справочник, который состоит из наиболее важных разделов. Здесь можно найти руководства и рекомендации по выполнению тех или иных операций, подробное описание методов исследования (работа с бактериями, бактериофагами, эукариотическими организмами, дигибридные системы, методы выделения и анализа ДНК про- и эукариотических организмов, работа с белками), методики и расчеты для приготовления растворов, подбор необходимых для исследования ферментов и реактивов. Можно следить за свежими публикациями. Имеются обзоры различных биологических ресурсов и программ, а также ссылки на биологические журналы и гранты биологического профиля. Внимание уделяется также образованию и образовательным ресурсам. Имеются сведения о компаниях и русскоязычных институтах биологического профиля, а также ссылки на полезные web-ресурсы.
nlm.nih.gov/
PubMed – это информационный ресурс Национального Института Здравоохранения США, состоящий из множества разделов. Он содержит более 16 миллионов цитат из научных журналов биомедицинской и естественнонаучной направленности, начиная с 1950-х годов. Здесь размещаются ссылки на полные тексты статей и другие связанные ресурсы (на страницы Национальной Библиотеки медицины США; на страницу Medline – базу материалов о более чем 700 заболеваниях и состояниях, о лекарственных средствах, на этом портале есть также медицинская энциклопедия и медицинский словарь и много другой полезной информации; на базы данных по токсикологии и токсическим веществам и др.). Поиск в базе данных журналов можно осуществлять по предмету или по названию журнала, по сокращенному названию, аббревиатуре ISO и другим параметрам.
ictprotein.org
PredictProtein является интернет-ресурсом для анализа последовательностей и предсказания структур и функций белков. Данный сервер позволяет произвести полный анализ первичной последовательности исследуемого белка, а также смоделировать вторичную и третичную структуры белков при использовании широкого набора разнообразных программ.
ссылка скрыта
Специализированная поисковая система для научных документов. Служба поисковой системы Google для поиска научных документов в зависимости от заданной предметной области. В принципе, достаточно быстрая и удобная поисковая система, с помощью которой можно найти научные статьи, публикации по интересующей пользователя предметной области.
ссылка скрыта
База данных и поисковая система, содержащая оглавления научных журналов издательства Elsevier по естественным наукам. Также, система содержит материалы по научной, медицинской и технической информации: более 2000 рецензируемых журналов, сотни книжных серий, руководств и справочников. Поиск информации осуществляется по ключевым словам. Возможен вариант расширенного поиска (по названию журнала, статьи).
ссылка скрыта/
Более 4000 ссылок на биологические и медицинские журналы содержится на "science.komm" (там же удобные ссылки на полнотекстовые источники, словари, базы данных по абстрактам и т.п.). По web-ссылке вы попадаете на сайт конкретного журнала. На многих журналах можно подписаться на рассылку оглавления по E-mail.
ссылка скрыта
Scirus – наиболее полная поисковая система для ученых в Интернете. Основанный на последних поисковых технологиях, он ищет более, чем в 300 миллионах определенных для науки Web-страницах, позволяя пользователям быстро находить:
- Научные, медицинские и технические сведения;
- Последние публикации; рецензируемые журналы; патенты и журналы, которые обычно пропускают другие поисковые системы.
- Поисковик предлагает уникальные функциональные возможности для ученых и исследователей
- Эта поисковая система обращает внимание только на те Web-страницы, которые содержат научную информацию.
Scirus поможет быстро определять местонахождение научной информации в Интернете:
- отфильтровывает ненаучные сайты;
- находит рецензируемые статьи формата PDF и файлы PostScript, которые являются часто невидимыми для других поисковиков;
- ищет глубже чем другие поисковые системы, показывая таким образом нужную информацию.
С Scirus, можно:
- выбрать диапазон предметных областей для поиска;
- сузить ваш поиск по конкретному автору, журналу или статье;
- ограничить поиск диапазоном даты;
- найти информацию о научных конференциях, резюме и патентах;
- усовершенствовать, настроить и сохранить результаты поиска.
l.expasy.org/
SWISS-model представляет собой полностью автоматизированный сервер, осуществляющий моделирование белковых структур по гомологии. Главная цель этого сервера — сделать моделирование белков, доступным для всех биохимиков и молекулярных биологов по всему миру.
ссылка скрыта
Сайт Высшей аттестационной комиссии Республики Беларусь, на котором размещены материалы, касающиеся подготовки научных кадров, присуждения ученых степеней и званий, краткие паспорта специальностей и программы-минимумы кандидатских экзаменов по специальности. В разделе «Каталог файлов» представлены доступные для скачивания файлы нормативных документов с приложениями и шаблоны регистрационных документов. Организован поиск по сайту и в сети Интернет.
ДЕЙСТВУЮЩИЙ ЛИЧНЫЙ САЙТ В WWW
ссылка скрыта
ГРАФ (КРУГ) НАУЧНЫХ ИНТЕРЕСОВ
Магистрантки Долгодилиной Елены биологический факультет
Специальность биохимия
| Смежные специальности
|