
Методика исследования площади поверхности адсорбента методика расчета энергии и энтропии адсорбционного взаимодействия Экспериментальные данные и их анализ
| Вид материала | Исследовательская работа |
- Методика расчета нормативных (технологических) потерь электроэнергии в электрических, 461.03kb.
- Методика расчета нормативных (технологических) потерь электроэнергии в электрических, 479.14kb.
- Методика исследования мотивации учения у первоклассников (Методика разработана в 1988г., 29.84kb.
- Методика расчета показателей эффективности инвестиционного проекта Методика анализ, 18.52kb.
- Методика «определение мотивов учения», 43.17kb.
- Енергетика та енергоресурсозбереження, 211.69kb.
- Методика расчета размера платы за услуги по передаче электрической энергии, 347.7kb.
- Методика исследования. 28 Методика компьютерной стабилографии. 28 Клиническая характеристика, 1222.25kb.
- Методика расчёта центробежного компрессора с радиальными лопатками, 253.33kb.
- ТемЫ рефератов по мерчендайзингу, 204.9kb.
Брянский городской лицей №1 имени А. С. Пушкина
Исследовательская работа
Изучение адсорбционных свойств -FeOOH
газохроматографическим методом
Выполнили: ученики 10 физико-химического класса
Брянского городского лицея №1
имени А. С. Пушкина
Ельцов А., Зелянин К., Корчагина К.
Научные руководители: кандидат химических наук,
преподаватель Брянского
государственного университета
Кузнецов С. В.,
учитель химии Брянского городского
лицея №1 Геращенков А. М.
Брянск 2007
ОГЛАВЛЕНИЕ
Стр.
Введение…………………………………………………………………………… 3
1. Литературный обзор…………………………………………………………… 4
1.1 Методы хроматографии. Газовая хроматография (ГХ)………………….... 4
1.2 Теория хроматографии и расчета хроматограмм ……………………....… 8
1.3 Адсорбенты и адсорбаты…………………………………………………… 16
2. Методика эксперимента и обработки данных……………….……………… 20
2.1 Получение γ-FeOOH……………………………………………………......... 20
2.2 Получение и очистка адсорбатов………………………………………......... 21
2.3 Методика хроматографии……………………………………………………... 22
2.4. Методика исследования площади поверхности адсорбента …………….... 23
2.5. Методика расчета энергии и энтропии адсорбционного взаимодействия... 24
3. Экспериментальные данные и их анализ……………………………………. 25
3.1 Алканы……………………………………………………………...………… 25
3.2 Ароматические соединения………………………………………………… 27
3.3 Гетероатомные соединения…………………………………………………. 28
Выводы…………………………………………………………………………… 31
Литература……………………………………………………………………….. 32
ВВЕДЕНИЕ
Известно, что адсорбция является не только предшествующей стадией гетерогенного взаимодействия, но и определяет тип, характер и условия этого взаимодействия. Исследования адсорбционных равновесий до настоящего времени являются актуальными. В связи не только с исследованиями по изучению каталитических свойств материалов или поиску новых катализаторов, но и ввиду необходимости уточнения теоретических вопросов адсорбции. Не менее важными являются вопросы адсорбции в коррозии и защите от нее.
Необходимо отметить, что -FeOOH являясь одним из основных продуктов коррозии, также используется для получения основного компонента магнитных носителей -Fe2O3. Этот процесс как правило осуществляется через стадию вакуумной дегидратации.
На поверхности оксидов и оксигидроксидов d-элементов имеются два основных типа групп: кислотно-основные (=FeOH2+,=FeOH,=FeO-) и группы донорно-акцепторного взаимодействия (свободные d-орбитали и не поделенные пары электронов кислорода).
В связи с выше сказанным представляется интересным исследовать вклад типов поверхностных групп -FeOOH во взаимодействия с различными адсорбатами.
Нами были поставлены следующие цели и задачи исследования:
1. Изучить адсорбцию следующих веществ:
а) неполярных растворителей: пропана, бутана, н-гептана, циклогексана;
б) полярных растворителей: бензола, пиридина, метилэтилкетона, диэтилового эфира.
в) углекислого газа.
2.Определить и сравнить значения теплот адсорбции, удерживаемого объема, времени удержания и энтропии для перечисленных выше веществ, при различных температурах с литературными данными по другим оксидам железа.
3.Исследовать вклад различных типов адсорбционных центров во взаимодействие с адсорбатами.
1. Литературный обзор
1.1 Методы хроматографии. Газовая хроматография (ГХ)
В период между 1950-1960 гг., т.е. в начальный период развития газовой хроматографии, её использовали для разделения не только многочисленных органических соединений, но и неорганических газов и простых летучих неорганических соединений, таких, как галогениды и гидриды, осуществляемого в основном методами газоадсорбционной хроматографии.
В начале 60-х годов постепенно начало возрастать значение распределительной газовой хроматографии, этому способствовало увеличение числа пригодных для ГХ неподвижных фаз. Большие надежды, которые возлагались в то время на ГХ как на метод элементного анализа, и в особенности как на метод анализа следов, оправдались лишь отчасти. Олигоэлементный анализ (анализ смесей, содержащих немного элементов) и многоэлементный анализ описаны лишь для нескольких случаев, имеющих практическое значение. При этом затруднения заключаются не в недостаточной разделительной способности или эффективности разделения, а в том, что возникают многочисленные проблемы, связанные с особенностями поведения сравнительно мало летучих неорганических соединений в газовой фазе и с взаимодействием этих соединений с материалом колонок и другими основными частями хроматографов [1]. Несмотря на эти затруднения, газохроматографическое разделение в сочетании с обнаружением чувствительными и селективными детекторами завоевало прочное место в анализе многих элементов, и особенно в анализе следовых количеств.
В органическом анализе задача ГХ заключается, главным образом в разделении сложных смесей веществ, причем обычно летучих, на индивидуальные компоненты. В тоже время в неорганическом анализе ГХ чаще используется для удаления веществ, мешающих анализу или для повышения селективности разделения путем специальных методов подготовки проб (получения производных и экстракции) и с целью подготовки проб для количественного анализа при помощи системы детекторов, расположенных за хроматографической колонкой.
Вообще говоря, применение хроматографического метода не ограничивается лишь разделением и анализом смеси веществ. В последнее время хроматография широко используется и как метод научного исследования растворов, газовых смесей и систем в условиях каталитических и др. взаимопревращений, исследования адсорбционных свойств твердых веществ, определения площади поверхности порошков их пористости, размера и объема пор [1].
Основными классификационными признаками являются:
- агрегатное состояние фаз;
- природа элементарного акта;
- способ относительного перемещения фаз;
- способ аппаратурного оформления процесса;
- цель осуществления процесса.
Классифицировать виды хроматографии можно на основе различных параметров (рис.1).
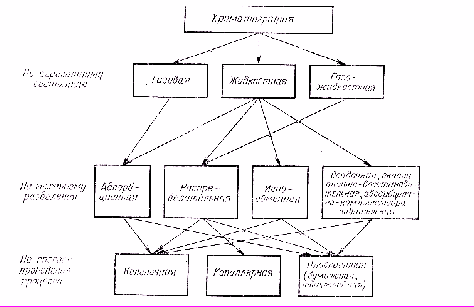
Рис. 1. Классификация хроматографических методов
Газовая хроматография – наиболее разработанный в аппаратурном оформлении хроматографический метод. Прибор для газохроматографического разделения и получения хроматограммы называется газовым хроматографом. Принципиальная схема газового хроматографа приведена на рис. 2.
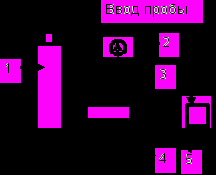
Рис. 2. Принципиальная схема хроматографа
Газ-носитель из баллона 1 непрерывно в течение всего опыта пропускается через всю систему: дозатор, колонку, детектор, измеритель скорости. Дозатор 2 служит для ввода в хроматографическую колонку 3 газообразной, жидкой или твердой пробы анализируемой смеси. В двух последних случаях смесь одновременно должна быть испарена. Разделение смеси на индивидуальные компоненты происходит в хроматографической колонке, которую характеризуют следующие параметры: L−длина колонки, или длина слоя сорбента, см; S−площадь поперечного сечения, см2; − доля свободного объема колонки, т. е. объема, занимаемого газом-носителем, включая и объем пор твердой фазы; 1–доля объема, занимаемого сорбентом (жидкой фазой для газо-жидкостной хроматографии и твердым адсорбентом для газо-адсорбционной хроматографии; для последней 1=(1 –); Vж−объем неподвижной жидкой фазы, см3; Vг–объем газовой фазы, см3; Vтв–объем твердого носителя (адсорбента), см3.
Vж =1LS;
Vг = LS.
Непосредственно на выходе газа из колонки устанавливается детектор 4, позволяющий зафиксировать изменение концентрации или потока вещества смеси по мере его вымывания из колонки (дифференциальный детектор) или же общее количество вымываемых компонентов (интегральный детектор). Современные детекторы имеют автоматическую запись результатов анализа, производимую самописцем 5.
На выходе из детектора газ-носитель с компонентами разделяемой смеси поступает в измеритель скорости потока (измеритель скорости не показан). Если объемная скорость, измеряемая жидкостным расходомером, равна р см3/мин, то для расчета истинного значения скорости на выходе из колонки следует учитывать давление насыщенного пара рабочей жидкости расходомера (обычно воды) Рв при температуре расходомера Тр. Тогда
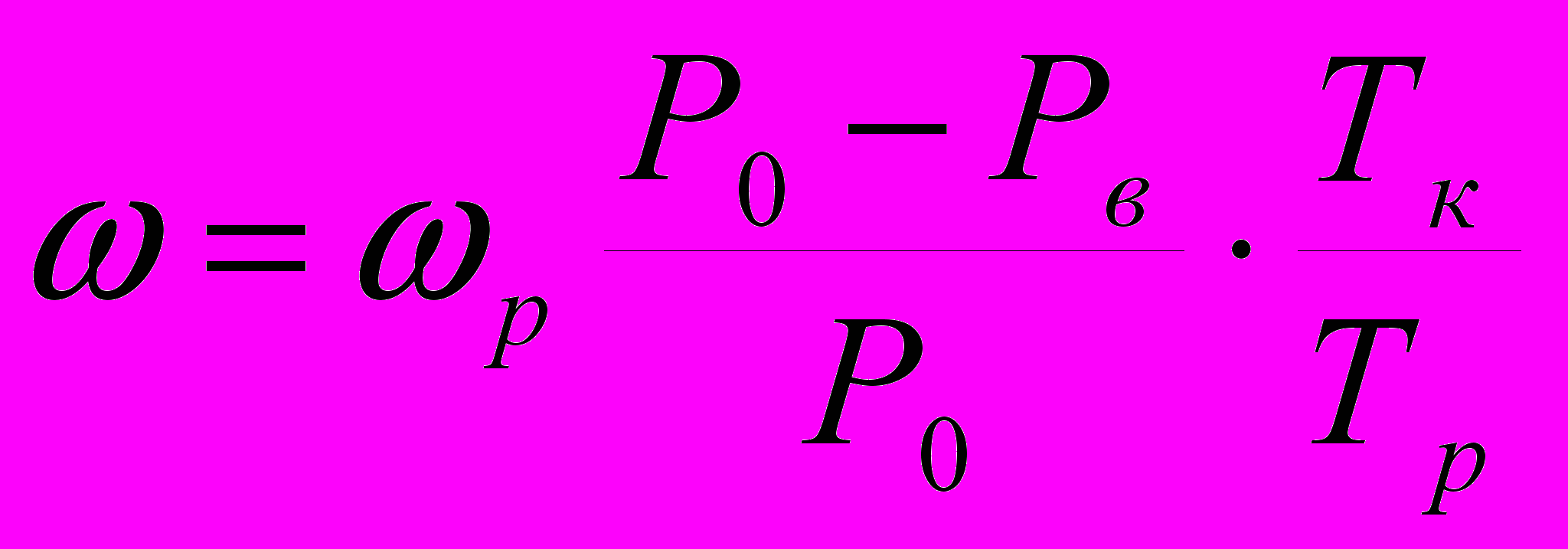
где Ро–давление газа на выходе из колонки; Тк–температура колонки. В ряде случаев расходомер устанавливается до колонки.
В основе газовой хроматографии лежат процессы адсорбции – десорбции или абсорбции – десорбции.
Адсорбцией называется процесс концентрирования вещества (адсорбата) поверхностью какого-либо твердого или жидкого вещества, называемого адсорбентом.
Абсорбцией называется избирательное поглощение вещества (абсорбата) объемом какого-либо твердого или жидкого вещества (абсорбента).
В общем случае процесс взаимодействия вещества (сорбата) с поверхностью или объемом другого вещества (сорбента) называется сорбцией. В газовой хроматографии может иметь место любой из процессов сорбции: адсорбция или абсорбция.
Процесс, обратный сорбции, получил название десорбции.
Хроматография - процесс, основанный на перемещении дискретной зоны вещества вдоль слоя сорбента в потоке подвижной фазы и связанный с многократным повторением адсорбционных и десорбционных актов.
1.2 Теория хроматографии и расчета хроматограмм
Впервые хроматографическое разделение сложной смеси (хлорофилла) было осуществлено М. С. Цветом в 1903 г.
При контакте с поверхностью неподвижной фазы каждый из компонентов разделяемой смеси распределяется между подвижной и неподвижной фазами в соответствии с его свойствами, например адсорбируемостью или растворимостью. Вследствие непрерывного движения подвижной фазы лишь часть распределяющегося компонента успевает вступить во взаимодействие с неподвижной фазой. Другая же его часть продвигается дальше в направлении потока и вступает во взаимодействие с другим участком поверхности неподвижной фазы. Поэтому распределение вещества между подвижной и неподвижной фазами происходит на небольшом слое неподвижной фазы только при достаточно медленном движении подвижной фазы. Поглощенные неподвижной фазой компоненты смеси не участвуют в перемещении подвижной фазы до тех пор, пока они не десорбируются и не будут снова перенесены в подвижную фазу [2]. Поэтому каждому из них для прохождения всего слоя неподвижной фазы в колонке потребуется большее время, чем для молекул подвижной фазы. Если молекулы разных компонентов разделяемой смеси обладают различной степенью сродства к неподвижной фазе (различной адсорбируемостью или растворимостью), то время пребывания их в этой фазе, а следовательно, и средняя скорость передвижения по колонке различны. При достаточной длине колонки это различие может привести к полному разделению смеси на составляющие ее компоненты.
Хроматографический процесс осуществляется при сорбционном распределении вещества между двумя фазами, одна из которых перемещается относительно другой.
Следует помнить, что состав смеси, покидающей хроматографическую колонку, непрерывно изменяется. В то время как в таких процессах, как экстракция или ректификация, можно отбирать в течение всего процесса непрерывно одну и ту же фракцию, или одно то же вещество, в хроматографическом процессе, за исключением специальных случаев, это делать нельзя.
Время удерживания и пропорциональный ему удерживаемый объем могут быть положены в основу качественной идентификации веществ в связи с тем, что эти величины определяются свойствами системы сорбат – сорбент.
Для качественной характеристики могут применяться как абсолютные, так и относительные величины удерживания. Однако применение абсолютных величин удерживания, например удельного удерживаемого объема Vм, может привести к получению ненадежных данных, так как при хроматографировании одних и тех же сорбатов на одинаковых сорбентах и в идентичных условиях на абсолютные величины удерживания могут оказать воздействие случайные факторы, например колебание температуры и т. п.
Применение относительных величин, например относительного удерживаемого объема, позволяет в значительной мере исключить действие случайных факторов и, следовательно, получать достаточно воспроизводимые результаты [3, 4]. В этом случае приведенный удерживаемый объем определяемого вещества Vм или приведенное время удерживания относят к приведенному удерживаемому объему Vм, стандартного вещества или к времени удерживания стандартного вещества, принятого за стандарт и полученного в одинаковых условиях. Однако область температур кипения анализируемых веществ может быть различна, поэтому в качестве стандартного соединения нельзя применять во всех случаях одно и то же вещество. Приходится удерживаемые объемы относить к различным стандартным веществам, объемы удерживания которых в общем случае не связаны между собой какими-либо соотношениями.
В газовой хроматографии, как правило, количественный анализ проводится не путем отбора отдельных порций анализируемого вещества на выходе из колонки, а по полученным на ленте самописца хроматограммам. Метод расчета количественного состава смеси зависит от типа применяемого детектора: дифференциального или интегрального. В хроматографическом анализе почти всегда применяются дифференциальные детекторы, поэтому мы рассмотрим только методы расчета по дифференциальным хроматограммам.
Дифференциальные хроматограммы состоят из ряда последовательно расположенных пиков, число которых должно соответствовать числу компонентов анализируемой смеси. Это условие не выполняется, если в хроматографической колонке не происходит разделения смеси двух или нескольких соседних компонентов. Взаимное наложение пиков нескольких компонентов может привести к ошибочному расчету количественного состава смеси. Поэтому очень важно предварительно проводить идентификацию всех компонентов смеси и знать ее полный качественный состав.
Вторым условием количественного анализа по хроматограммам является полное поступление в колонку всех компонентов введенной для анализа пробы и, следовательно, полное ее испарение. Наконец, экспериментатор должен быть уверен в том, что ни одно из анализируемых веществ не вступает в необратимую реакцию с сорбентом или твердым носителем, а также в том, что все вещества регистрируются детектором.
В случае необходимости полной расшифровки состава анализируемой смеси все ее компоненты должны быть разделены, хотя существуют приближенные методы количественного расчета для неполного разделения смеси веществ. Желательно, чтобы форма пика на хроматограмме была симметричной, как можно ближе отвечающей гауссовской кривой распределения. Формы получаемых пиков зависят от формы изотермы адсорбции. На рис. 3 представлены формы изотерм и соответствующие им хроматографические пики.
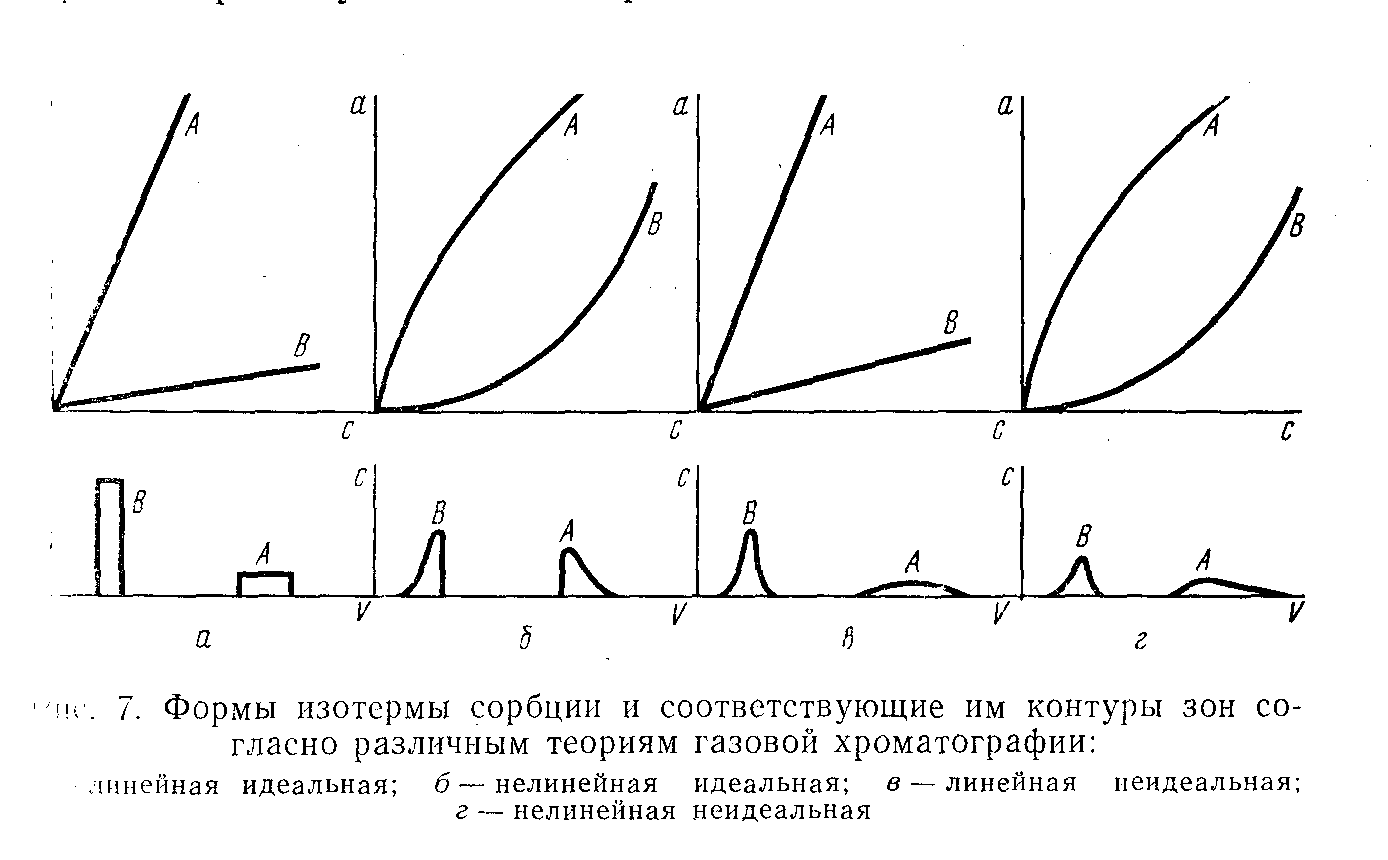
Рис . 3. Формы изотерм и соответствующие им хроматографические пики
В основу количественного определения состава анализируемой смеси по дифференциальным хроматограммам можно положить высоту пиков h, площадь пика П или же произведение высоты пика h на величину пропорциональную (рис. 4).
Площадь пика для концентрационных и потоковых детекторов пропорциональна количеству введенного в колонку вещества.
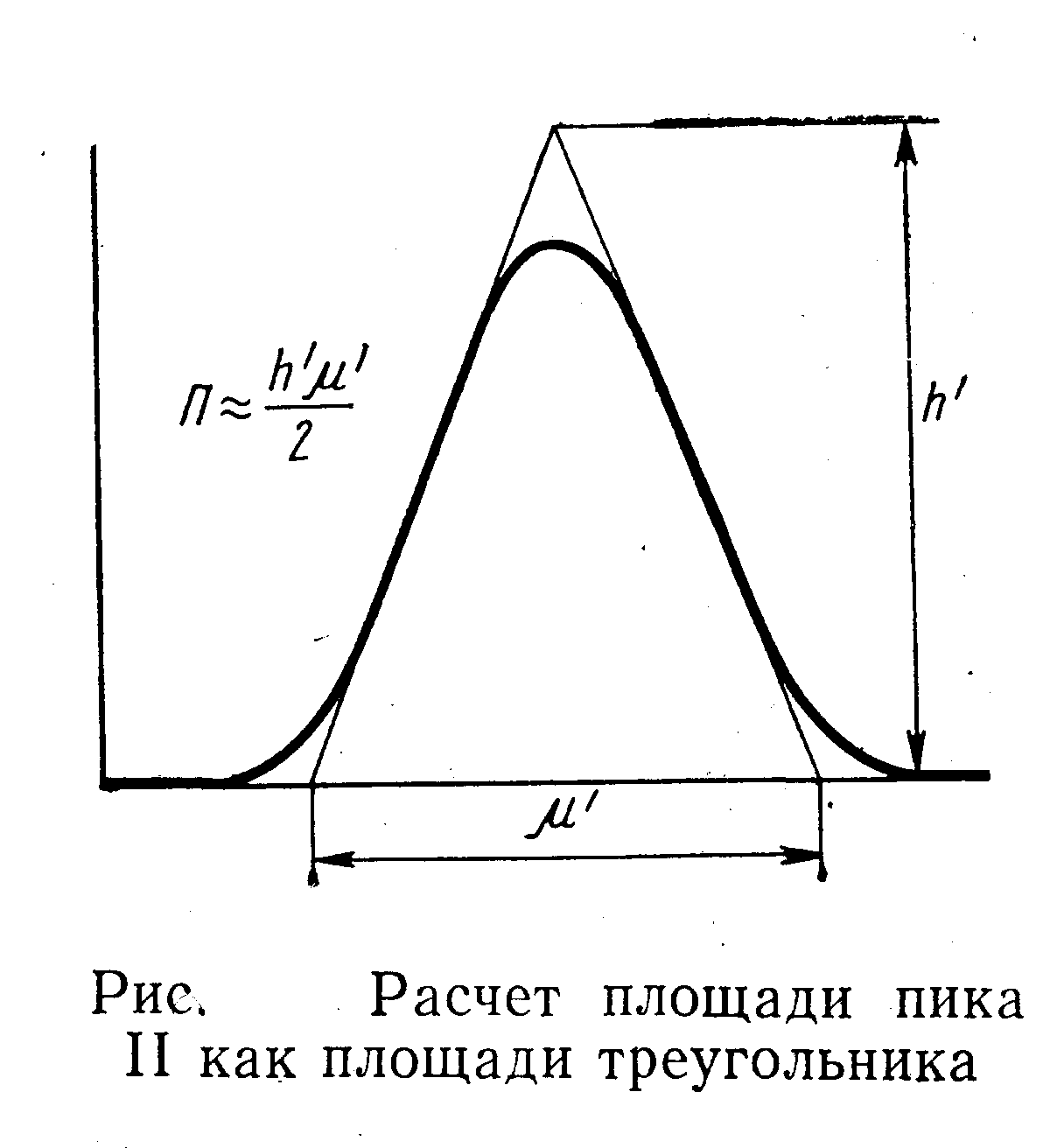
Рис. 4. Обозначение и правила измерения величин
при количественном расчете по хроматографическому пику
Высоту хроматографических пиков измеряют различными способами, но наиболее точные приведены на рис. 5 для различных форм и типов наложений пиков.

Рис. 5. Способы измерения высоты пиков по базовой линии
Концентрацию вещества, соответствующую максимуму зоны, можно определить по уравнению:
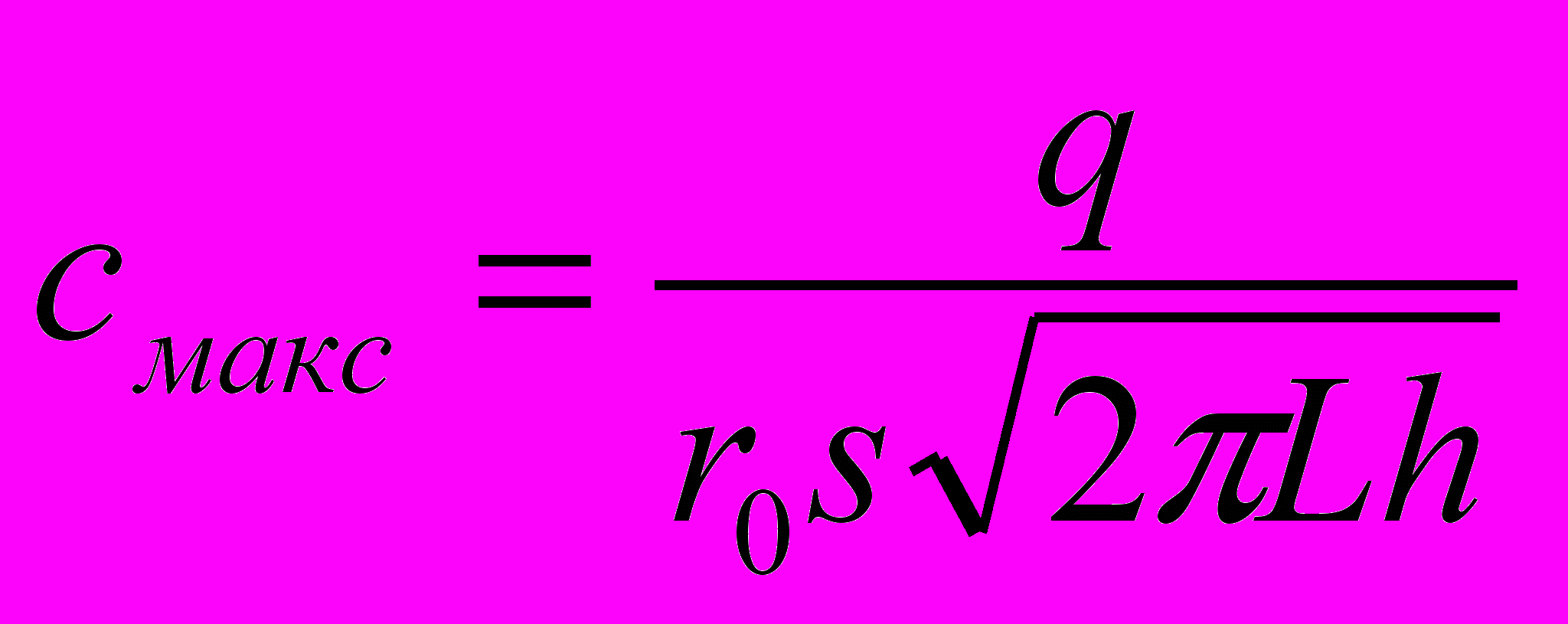
Следовательно, Смакc пропорциональна количеству введенного вещества, а значит, и h пропорциональна . Отсюда следует, что так как площадь пика
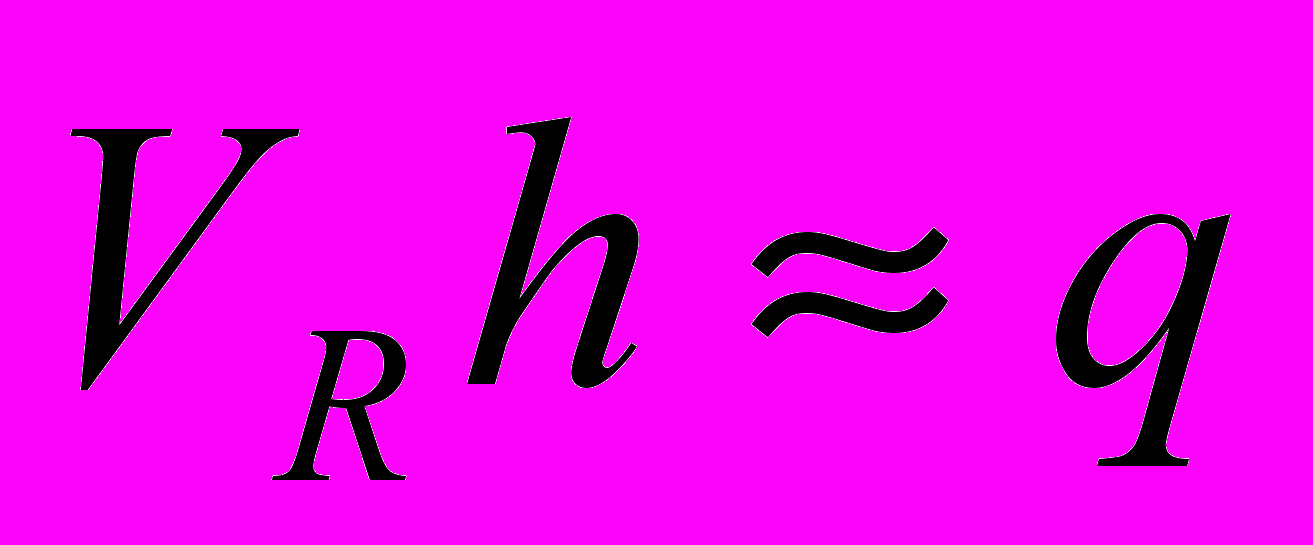 и
и  , то
, то
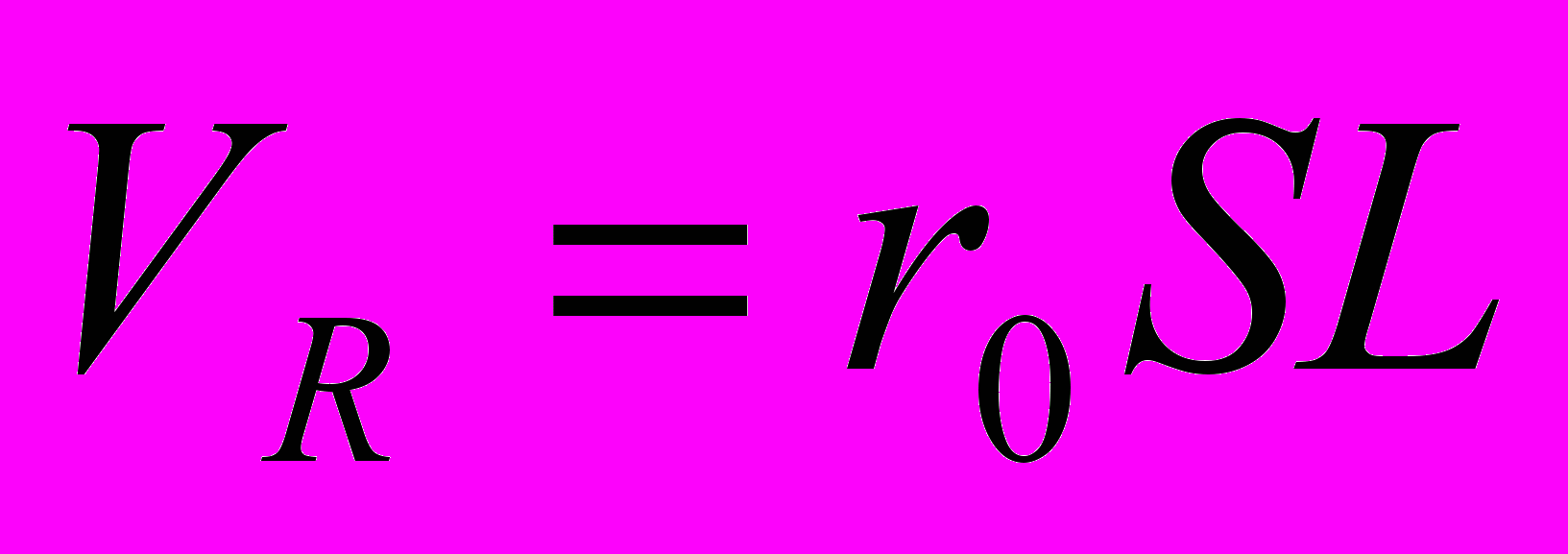

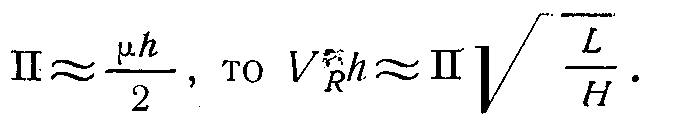

Итак, для количественной характеристики могут служить как высота пика h, так и его площадь П. Выбор для расчета того или иного параметра зависит, главным образом, от формы хроматографических пиков и их взаимного расположения.
Измерение хроматографических пиков может привести к существенным погрешностям и оказать влияние на точность количественного анализа. Поэтому ко всем методам измерения пиков предъявляются следующие требования: они должны обеспечивать хорошую воспроизводимость измерений даже в случае частичного наложения пиков; каждое измерение должно выполняться не менее 3–5 раз с тем, чтобы иметь возможность пользоваться средними величинами; измеряемые величины должны линейно зависеть от концентрации или потока вещества во всем диапазоне измерений.
Наиболее простым является измерение высоты пика. Однако для пиков размытых и не имеющих острой вершины измерение высоты может привести к значительным погрешностям.
Площадь пика определяют планиметром либо взвешиванием на аналитических весах вырезанного из ленты самописца хроматографического пика и нормализацией пиков по массам. Для симметричных пиков целесообразно измерять площадь как произведение высоты на половину ширины. В этом случае за высоту следует принимать отрезок h', а за ширину–отрезок .' (рис. 4). Измерение площади для очень острых и узких пиков может привести к существенным ошибкам. Величину удержания измеряют по хроматограмме как расстояние от момента выхода несорбирующегося вещества до проекции точки максимальной концентрации на нулевую линию.
Измерение высоты пиков более быстрый метод, чем расчет площадей. Однако диапазон линейной связи высоты пика с величиной вводимой пробы по сравнению с такой связью для площадей пиков меньше. Поэтому измерения по высотам пиков производят в тех случаях, когда количество вводимой пробы не превышает 100 мкг для насадочных колонок и 0,1 мкг – для капиллярных. Если наблюдается дрейф нулевой линии, то измерение высоты производят так, как показано на рис. 5 для пиков веществ О и С.
Площади пиков в меньшей степени зависят от условий эксперимента, кроме того, измерение площадей дает большую точность. По-видимому, эти обстоятельства явились причиной преимущественного распространения количественных измерений по площадям, чем по высотам пиков.
Измерение площадей при помощи планиметра весьма трудоемко и менее точно, чем другие методы. Кроме того, наблюдается плохая воспроизводимость результатов. Хороших результатов можно добиться при вычислении площади умножением высоты на ширину пика, соответствующую половине высоты. Этот метод быстр и точен, кроме того, точность измерения можно повысить, увеличивая скорость протяжки диаграммной ленты.
Все три параметра могут применяться для количественного расчета одним из следующих принятых в хроматографии методов; нормировкой, нормировкой с калибровочными коэффициентами, внутренней стандартизацией и абсолютной калибровкой.
Метод нормировки. Этим методом определяют соотношение между количеством анализируемых компонентов смеси и одним из параметров хроматограммы, например площадями пиков. При этом сумма одного из параметров всех пиков принимается за 100%. Тогда содержание 1-го компонента в смеси (в процентах) определяется по формуле:

При этом все компоненты должны регистрироваться на хроматограмме.
Метод нормировки очень удобен, так как не требует знания, а следовательно, и точности дозировки вводимой пробы анализируемой смеси. Однако он применим только в том случае, если чувствительность детектора одинакова для всех детектируемых веществ. Практически это требование осуществляется очень редко. Поэтому метод нормировки следует применять с большой осторожностью даже при анализе смеси веществ близкого строения.
Метод нормировки с калибровочными коэффициентами. В этом методе, как и в методе нормировки, принимают сумму одного из параметров за 100% и относят к ней параметр каждого из компонентов смеси. Однако для учета различия в чувствительности детектора предварительно для каждого компонента находят поправочные коэффициенты. Калибровку проводят так, что один из постоянно присутствующих и обычно преобладающих компонентов смеси считают сравнительным и значение его поправочного коэффициента принимают за 1. Тогда калибровочные коэффициенты других компонентов смеси рассчитывают, измерив соответствующие параметры, по формулам:
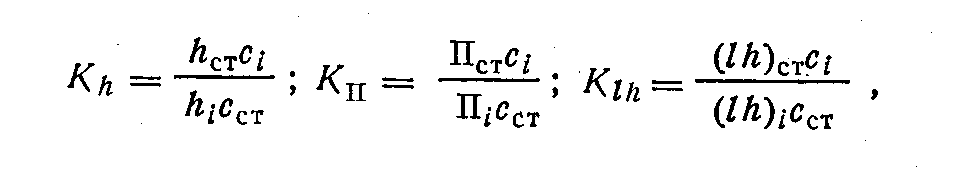
где индекс «ст» относится к веществу, принятому за сравнительное, а индекс i — к искомому компоненту.
Определенные таким образом калибровочные коэффициенты можно применять для расчета процентного состава смеси. Например, если расчет производится по площадям пиков, то процентное содержание i-го компонента в смеси можно рассчитать по формуле:

В зависимости от метода калибровки поправочные коэффициенты могут быть отнесены к массовым, объемным или мольным долям вещества. Для взаимного пересчета поправочных коэффициентов полезно знать соотношение:

где Мi и Мст – молекулярные массы анализируемого вещества и стандарта.
Достоинства метода нормировки с поправочными коэффициентами заключаются в отсутствии необходимости точного измерения количества вводимой пробы, а также в применении относительных поправочных коэффициентов, что снижает действие небольших колебаний эксперимента на точность измерений.
К недостаткам метода следует отнести необходимость идентификации каждого пика, неприменимость метода для анализа, если в смеси присутствует хотя бы один не регистрируемый детектором компонент, а также то, что ошибки в определении одного компонента вызывают искажение результатов всего анализа.
Несмотря на эти недостатки, метод нормировки с поправочными коэффициентами широко применяется в хроматографическом анализе. Это объясняется хорошей воспроизводимостью результатов, отсутствием необходимости точной дозировки пробы, а также возможностью обходиться без чистых стандартных веществ и наличием данных о поправочных коэффициентах в справочной литературе [4].
Возможные ошибки при расчете по хроматограммам. При количественном анализе по хроматограммам следует учитывать кроме обычных постоянных, систематических и случайных ошибок переменную систематическую ошибку, которая может возникать в связи с нерегистрируемым на хроматограмме соединением. Отсутствие регистрации компонента детектором может быть связано с невыходом высококипящего соединения из колонки, с конденсацией вещества до детектора, нечувствительностью детектора либо с другими причинами. Какова бы ни была причина переменной систематической ошибки, она должна быть тем или иным способом устранена.
Ошибки в определении количественного состава по хроматограммам в значительной степени определяются погрешностью при расчете хроматографических пиков. Поэтому важно, чтобы при этих измерениях выполнялись следующие основные требования:
1) должна быть хорошая воспроизводимость измерений;
2) результаты измерений должны линейно изменяться с изменением концентрации определяемого вещества;
3) метод измерения должен быть достаточно простым.
- Адсорбенты и адсорбаты
Между молекулами адсорбируемого вещества и адсорбента существует взаимодействие определенного вида, зависящее от природы как газообразного вещества, так и адсорбента. Простейшим случаем является адсорбция неполярных молекул газа на поверхности неполярного же адсорбента. При таком виде адсорбции действуют лишь дисперсионные силы притяжения и силы отталкивания.
Если адсорбент построен не из атомов, а из ионов, то к дисперсионным силам притяжения добавляются индукционные силы притяжения диполя, индуцированного в молекуле адсорбата электростатическим полем от ионов решетки адсорбента. Доля индукционных сил в величине потенциальной энергии адсорбции пропорциональна поляризуемости молекулы адсорбата и квадрату напряженности электростатического поля над поверхностью адсорбента. Напряженность же зависит от заряда иона, типа решетки и грани.
При адсорбции полярных молекул на неполярном адсорбенте постоянный дипольный момент молекулы адсорбата поляризует атомы адсорбента, т. е. индуцирует в них электрические моменты. В результате возникает индукционное притяжение, накладываемое на дисперсионное.
Адсорбция полярных молекул на адсорбенте, имеющем ионы или диполи, вызывает взаимодействие диполя адсорбата с электростатическим полем адсорбента. Если молекулы адсорбата невелики и обладают периферийно расположенными диполями, как например, у молекул воды или аммиака, то они ориентируются в электростатическом поле адсорбента. При этом возникает ориентационное кулоновское взаимодействие.
При адсорбции на адсорбентах, содержащих на поверхности гидроксильные группы, возникает ассоциация молекул адсорбатa вызванная образованием водородной связи. В этом случае общая энергия взаимодействия адсорбата с адсорбентом увеличивается. В результате теплота адсорбции веществ, образующих водородную связь с адсорбентом, оказывается больше теплоты адсорбции веществ, сходных по геометрической форме и близких по величине энергии дисперсионного притяжения, но не образующих водородной связи.
Образование водородной связи наиболее характерно для адсорбентов типа гидроксилированных силикагелей, алюмогелей, алюмосиликатных катализаторов и т. п. На поверхности этого рода адсорбентов легко адсорбируются вещества, способные к образованию водородных связей, такие как вода, спирты, аммиак, амины и др. Например, при адсорбции этих веществ на гидроксилированной поверхности кремнезема водородные связи могут образовываться по нижеследующим схемам:

Взаимодействие функциональных групп молекул адсорбата с гидроксильными группами поверхности адсорбента увеличивает энергию адсорбции молекул, имеющих дипольные и квадрупольные моменты или -электронные связи, и мало изменяет энергию адсорбции молекул с симметричными электронными оболочками. Следовательно, если удалить с поверхности адсорбента гидроксильные группы, то снизится адсорбция адсорбата, молекулы которого имеют дипольные и квадрупольные моменты или -электронные связи, и мало изменится активность адсорбента для соединений с симметричными электронными оболочками.
В связи с изложенным представляет интерес замена активных гидроксильных групп поверхности адсорбента на неактивные путем обработки адсорбента соответствующим образом подобранными химическими реагентами. Такое изменение поверхности адсорбента носит название химической модификации. Например, активные группы ОН на поверхности кремнезема можно заменить на неактивные триметилсилоксильные группы обработкой триметилхлорсиланом. Изменение активности поверхностных групп может происходить под действием как температурных градиентов, так и в результате окислительно-восстановительных процессов. [5].
Такое изменение химической природы поверхности адсорбента снижает ее активность и энергию взаимодействия с адсорбатом, что имеет большое значение для изменения свойств адсорбата в желаемом направлении.
Выбирая для практических целей адсорбент, следует учитывать как грубую геометрическую структуру, так и химическую природу. К первой относятся удельная поверхность, степень пористости, диаметр пор, степень дисперсности, к последней − химическая структура и химическая активность поверхности адсорбента, наличие атомарной или ионной решетки и др.
Большое влияние на адсорбцию, а также на избирательные свойства адсорбентов оказывают размеры их пор. Так, благодаря малым размерам пор некоторые адсорбенты могут оказаться недоступными для крупных молекул. В порах с небольшим диаметром могут относительно легче проявляться процессы капиллярной конденсации. Все это позволяет сделать вывод о необходимости классифицировать адсорбенты по их геометрической структуре. Такая классификация предложена Киселевым 1949 г. [6].
I тип – непористые адсорбенты. Сюда относятся моно- и поликристаллические вещества, такие, как графитированная сажа, хлорид натрия, а также аморфные непористые вещества. Удельная поверхность подобных адсорбентов может колебаться в широких пределах −от сотых долей до сотен квадратных метров на грамм. Характерна для этого типа независимость адсорбционных свойств единицы поверхности от удельной поверхности.
II тип − однородноширокопористые адсорбенты. Сюда относят широкопористые ксерогели, крупнопористые стекла и спрессованные в таблетки порошки непористых адсорбентов. Для этого типа характерна высокая удельная поверхность (порядка сотен квадратных метров на грамм) и значительные размеры пор (100—200 А). Капиллярная конденсация может происходить достаточно легко, причем характерен капиллярно-конденсационный гистерезис.
ΙΙΙ тип − однороднотонкопористые адсорбенты. Сюда относят тонкопористые ксерогели, тонкопористые стекла, активные угли, а также пористые кристаллы, в том числе цеолиты типа А и X. Эти последние ведут себя как молекулярные сита.
IV тип − неоднороднопористые адсорбенты. К ним относят ксерогели, получаемые осаждением гидрогелей из растворов силикатов солями сильных кислот. В них много сильно адсорбирующих тонких пор различного диаметра.
Адсорбентом может быть любое вещество. Но в зависимости от природы оно по разному ведет себя по отношению к одному и тому же адсорбату [7]. На тип адсорбционного взаимодействия влияет как предыстория адсорбента [8], так и наличие уже адсорбированных или соотсорбирующихся соединений [9].
Известно [6], что поверхность всех оксидов (проводников, полупроводников и диэлектриков) покрыта гидроксильными группами. В работе [6] показано, что кислотно-основные центры не могут быть полностью удалены с поверхности даже при нагревании до 600-1000С.
Кислотно-основные свойства поверхности оксидов– это одна из фундаментальных характеристик, которая регулирует протекание равновесных процессов на границе адсорбент/адсорбат. В это понятие включается сила кислотных(основных) центров, которая характеризуется обычно функцией Гаммета (Н0) и константы кислотно-основных равновесий (рКi). По типу взаимодействия их делят на кислотно-основные и донорно-акцепторные центры.
В последние годы в ряде работ предприняты исследования по разделению и выявлению типов кислотно-основных центров.
Особенность кислотно-основных центров на оксиде является их высокая активность. По силе поверхностных кислот и оснований они приближаются к сверхкислотам и сверхоснованиям (сверхкислоты – вещества, обладающие более высокой кислотностью, чем 100% серная кислота).
2. Методика эксперимента и обработки данных
2.1 Получение γ-FeOOH
В основе синтеза лежала методика, указанная в [10]. Для удаления железа (III) раствор хлорида железа (II) перед опытом выдерживался с небольшим избытком соляной кислоты над порошком карбонильного железа до прекращения выделения водорода (одновременно раствор продувался аргоном). После определения концентрации железа раствор FeCl2 смешивался с обескислороженными растворами уротропина и нитрита натрия. Нами установлено, что за исключением избытка уротропина отклонение от прописи (по Брауэру) при проведении синтеза мало влияет на чистоту продукта. Проведение синтеза с сокращением времени нагревания до 30 мин и последующим медленным охлаждением приводит к получению более светлого практически не слипающегося продукта (однако выход продукта был примерно 20%). Нами была использована методика, позволяющая получать монодисперсный продукт высокой чистоты [11].
После смешивания раствор помещался в центрифужные стаканы и при постоянной частоте вращения выдерживался 10 минут. Через каждые 10 минут раствор сливался с осадка, осадок поступал на промывку, а раствор использовался далее для последующего осаждения (процесс получения не превышал 2 – 2,5ч). Отмывка осадка проводилась с использованием центрифуги при 7500 об/мин по 20 минут до отрицательной реакции на Cl-. Размер частиц зависит от частоты вращения при данном размере ротора. Это позволяет прогнозировать размер получаемых частиц.
- Получение и очистка адсорбатов
В работе использовались реактивы как заводского изготовления, так и полученные непосредственно в лаборатории.
Гептан «нормальный» квалификации для хроматографии.
Бензол, пиридин, метилэтилкетон, ацетон, диэтиловый эфир квалификации «ЧДА» или «ХЧ».
Диэтиловый эфир подвергался дополнительной очистке. Эфир встряхивался с подкисленным (Н2SO4) раствором сульфата железа(II). Порции сульфата железа(II) сменялись до тех пор, пока водный раствор KI с пробой диэтилового эфира не переставал давать оранжевого окрашивания (выделение иода). Далее следовала перегонка на металлическим натрием.
Пропан и бутан получали электролитическим восстановлением диметилкетона и метилэтилкетона соответственно. В U-образную трубку помещали 25 раствор серной кислоты, в который вводилось 5-10 диметилкетона или метилэтилкетона. Данный раствор подвергался электролизу с цинковым амальгамированным катодом. По литературным данным [12] на таком электроде происходит 100 восстановление кетонов или спиртов в соответствующие углеводороды. Газообразную фазу пропускали через холодильник, где конденсировалась основная масса паров воды, ацетона и метилэтилкетона. Далее газовая смесь пропускалась через колонку с активированным углем и силикагелем, для полной очистки и осушки. Очищенные пропан или бутан замораживались жидким азотом (для удаления водорода) и испарялись в резиновые груши для хранения и отбора проб. Углекислый газ получали из карбоната кальция при взаимодействии с соляной кислотой. Газ промывался водой и осушался прокаленным хлоридом кальция. Используемый в качестве не адсорбируемого компонента водород, получали электролизом по методике указанной в [13].
2.3. Методика хроматографии
У
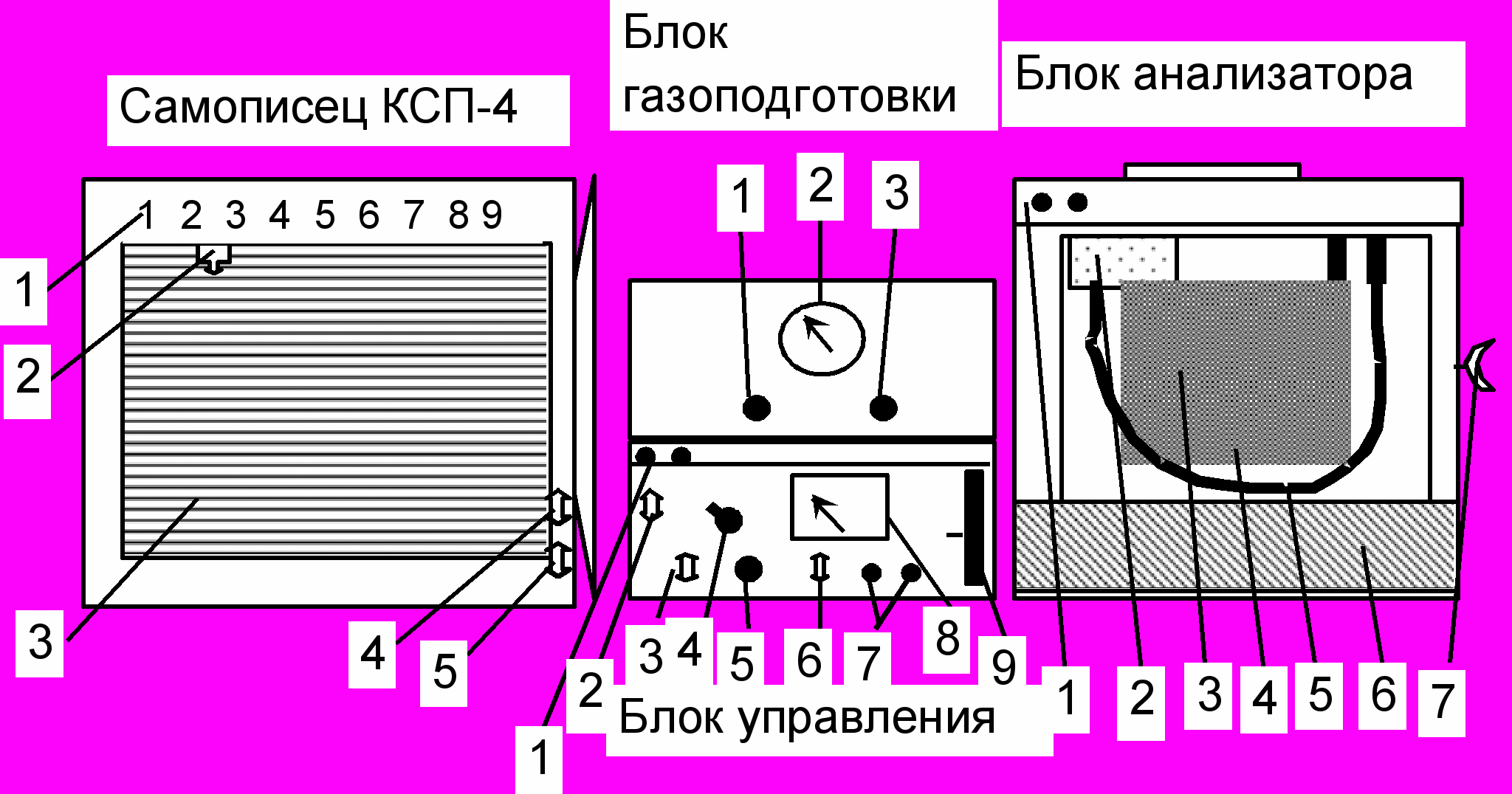
стройство и основные узлы хроматографа ЛХМ – 8МД (1) представлены на рис.6.
Рис. 6. Схема расположения основных элементов управления хроматографом ЛХМ – 8МД (1)
Самописец (вид с открытой дверцей): 1-шкала прибора, 2-пишущее устройство, 3-диаграммная лента, 4- тумблер включения движения диаграммной ленты, 5- тумблер «сеть».
Блок газоподготовки: 1- регулятор скорости потока газа в колонке №1, 2- манометр входного давления, 3- регулятор скорости потока газа в колонке №2.
Блок управления: 1- панель с сигнальными лампами включения и нагрева термостата в блоке анализатора.
При работе на хроматографе в качестве газов носителей использовали гелий и азот высокой чистоты (дополнительной очистке не подвергались). В качестве не удерживаемого компонента использовали водород, полученный электролизом раствора щелочи.
Жидкие пробы вкалывались хроматографическим микрошприцем в колонку (объем пробы 1,5-8 мкл). Газообразные соединения вводились в колонку через встроенный кран дозатор с объемом петли 0,25 – 5 мл.
2.4. Методика исследования площади поверхности адсорбента
Определение удельной поверхности адсорбента по хроматографическим данным [14]. В этом методе определяют удельный удерживаемый объем, хроматографируя какой-либо газ на адсорбенте, удельная поверхность которого известна. За τg,l = τR- τRм тем определяют удельный удерживаемый объем, хроматографируя тот же газ и в тех же условиях, но на другом адсорбенте, поверхность которого требуется определить. Обязательным условием является, что физические и химические характеристики эталонного и изучаемого адсорбентов близки. Тогда поверхность исследуемого адсорбента может быть рассчитана по формуле (S2A=
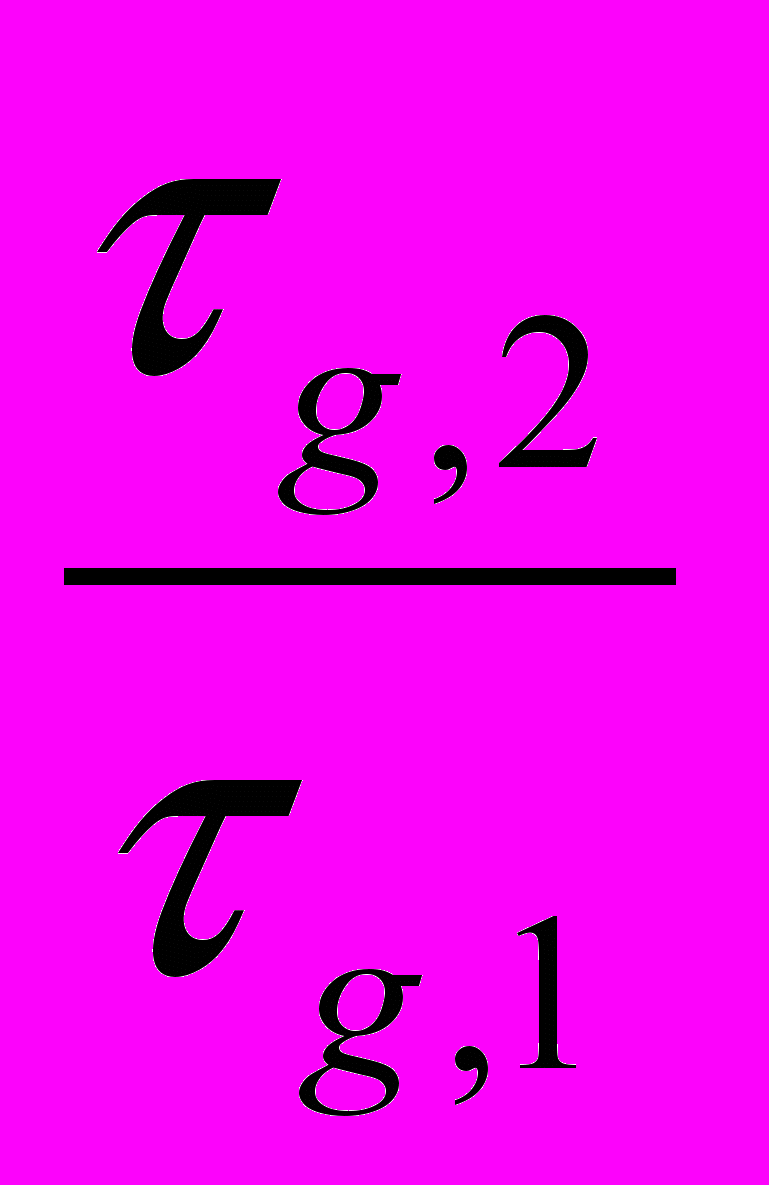 S1A).
S1A).Колонку хроматографа длиной не более 1 м заполняют взвешенным количеством адсорбента, удельная поверхность которого. Присоединяют колонку к хроматографу. Продувают систему чистым азотом, устанавливают требуемую скорость его потока и добиваются постоянства нулевой линии на ленте самописца. Вводят в колонку хроматографа 5 мл этана или этилена. В момент ввода включают секундомер. Наблюдают за пером самописца. Когда оно достигнет максимального отклонения, секундомер останавливают. Полученная величина представляет собой время удержания τR, пропорциональное удерживаемому объему VR. Поэтому, вычисляя удельную поверхность, нет необходимости переводить τR в VR . Измерения повторяют 5-6 раз. Для расчета принимают среднее арифметическое значение. Из полученной величины τR требуется вычесть τRм- время удержания неадсорбирующегося газа, например водорода. Измеряют τRм так же как и τR, вводя в колонку 5 мл водорода. Величину τg для расчета получают по формуле:
τg,l =

где
 - навеска адсорбента.
- навеска адсорбента.Получив значение τg,l для известного адсорбента, заменяют в колонке хроматографа адсорбент на исследуемый и повторяют все измерения при той же температуре, рассчитывая затем значение τg,2.
Удельную поверхность адсорбента рассчитывают по формуле:
S2A=
S1A2.5. Методика расчета энергии и энтропии адсорбционного взаимодействия
По усредненным исправленным удерживаемым объемам [15] рассчитывались с теплоты адсорбции, изменение энтропии адсорбционного взаимодействия. Рассчитанные величины отнесены к массе адсорбента.
Для расчета использовались следующие уравнения:
lgVМ=U/RT+B , где lgVМ – логарифм исправленного удерживаемого объема отнесенный к единице массы адсорбента (г), U = - q – дифференциальная теплота адсорбции, R и Т – газовая постоянная и температура (К) соответственно, В – свободный член, показывающий «теплоту адсорбции при абсолютном нуле».
-F=RTIn Vм , где -F – величина свободной энергии.
S = (F — U)/Т, где S – величина изменения энтропии.
3. Экспериментальные данные и их анализ
3.1 Алканы
Для пропана и бутана время удержания не зависят от объема введенной пробы. Изменение теплоты адсорбции с увеличением температуры, а также значения теплоты и энтропии адсорбции находятся в достаточно хорошем соответствии с литературными данными [15] (1,5-3 кДж). Для бутана при высокой температуре (100-120С) наблюдается уменьшение теплоты адсорбции. Для данного факта объяснения пока не найдено, возможно он связан с приборной ошибкой. Форма хроматографических пиков для бутана и пропана симметричная, с малой шириной. Ширина пиков практически не меняется с температурой. Это еще раз указывает на низкую энергию взаимодействия. Независимость времени удержания от объема введенной пробы указывает на отсутствие пористости сравнимой с размерами указанных газов. Малое время удержания не позволяет использовать данные газы для определения площади поверхности исследуемого адсорбента сравнительным методом. Оцененная величина площади лежит в интервале 40-120 м2/г.
Полученные хроматограммы н-гептана рис.7, в противоположность пропану и бутану, обнаруживают, зависимость времени удержания, как от объема введенной пробы так и от времени между вводами проб. Форма пиков не симметричная. Как видно из рисунка время удержания уменьшается с увеличением объема введенной пробы. С увеличением температуры при постоянном объеме время удержания уменьшается рис.8. Высокое время удержания гептана мы связываем с размерным фактором поверхности, в образце присутствуют поры сравнимых размеров с молекулой гептана. Резкое уменьшение времени удержания с повышением температуры для гептана можно связать с конформационными изменениями молекулы. Для гептана объем пробы 0,5 мкл. не дает хроматографического отклика. Объем пробы 0.5-2 мкл. дает растянутый несимметричный пик с большим временем удержания.
При увеличении пробы до 6-8 мкл. время удержания не меняется, форма пика приобретает более симметричный вид. Но зависимость времени удержания от температуры более явно проявляется с уменьшением пробы.
Полученные данные позволяют оценить объем микропор исходя из размеров молекулы гептана в различных конформационных изменениях. В низкотемпературной области время удержания гептана слабо зависит от температуры, что может, определяется конформационным фактором (энергия конформационных изменений находится в пределах 50-70 кДж/моль).

Рис. 7. Зависимость формы хроматографических пиков гептана от объема пробы: температура 70С; 1 - 7мкл, 2 - 5 мкл, 3 - 3 мкл.
Теплота адсорбции при температуре 80С равна 30 кДж. Для точной размерной оценки необходимо провести измерения с циклогексаном.

Рис. 8. Логарифм удерживаемого объема от обратной температуры.
Объем пробы 2 мкл. 1-метилэтилкетон, 2-пиридин, 3-диэтиловый эфир, 4-гептан, 5-бензол.
3.2. Ароматические соединения
Наличие высокой π-электронной плотности в ядре бензола и гомологов должно приводить к проявлению более сильных донорно-акцепторных взаимодействий с незаполненной d-оболочкой ионов железа, при этом бензол выступает в качестве донора электронной плотности, а ионы железа в роли акцептора.
Специфичность такого взаимодействия наглядно демонстрируется s-образной формой кривой на кривой рис.8. Наряду с бензолом делались попытки использования толуола и ксилола, но в указанном температурном интервале хроматографический отклик получен не был. Повышение температуры до 180С дает хороший хроматографический отклик, но при таких температурах происходит превращение -FeOОН в -Fe2O3. Поэтому от использования толуола и ксилола в качестве адсорбатов было решено отказаться.
Исследования с бензолом показывают несколько иную зависимость чем с гептаном (рис.7-9). Для бензола объем пробы 0,5 мкл. не дает хроматографического отклика, а большие объемы дают более или менее ровные пропорциональные пики.
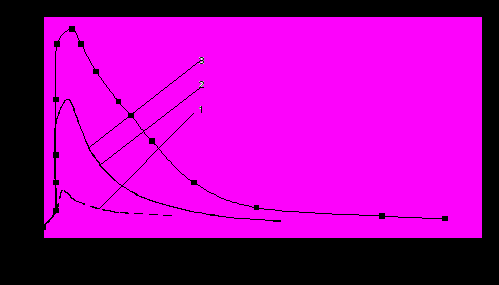
Рис. 9. Зависимость формы хроматографических пиков бензола от температуры. Объем пробы 5 мкл; 1 - 50С , 2 - 70С, 3 - 90С
Аналогичная зависимость наблюдалась в [16] и объясняется изменением числа центров адсорбции с температурой и их температурной активацией. Изменение формы пиков с увеличением температуры связано с изменением типа адсорбции или перестройкой поверхностных гидроксильных групп экранирующих адсорбционные центры. Небольшая разница в теплоте адсорбции бензола и этилметилкетона (как будет показано ниже) подтверждает предположение об экранировании d-электрофильных центров, образованных ионами железа, поверхностными гидроксогруппами. Из отношения массы навески к площади и объема введенной пробы (бензол, пиридин) до уровня начала ее выхода из колонки позволяет оценить число адсорбционных центров на поверхности (5*10(16-18) м-2). Полученная величина превышает на несколько порядков величину центров FеО(Н)2+ и FеОН полученную в работе[17]. Подобное расхождение хорошо согласуется с предположением о том, что для ароматических соединений гидроксильные поверхностные группы не являются адсорбционными центрами. Полученная выше величина числа адсорбционных центров близка к числу атомов железа находящихся на поверхности оксида.
3.3. Гетероатомные соединения
Использование следующих соединений прежде всего связано с технологическим их применением (за исключением пиридина) и во вторую очередь их полярностью. Время удержания диэтилового эфира прямо пропорционально изменяется с температурой рис.10, время удержания не зависит от введенного объема. Для метилэтилкетона и диметилкетона линейная зависимость от температуры не наблюдается. Значительной разницы между диметилкетоном и ацетоном нами не получено, очевидно, это связано с малой разницей дипольных моментов этих соединений.
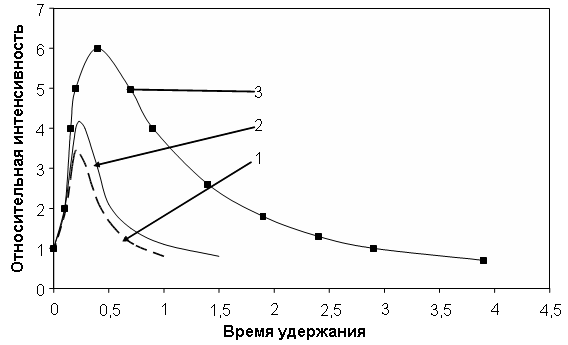
Рис. 10. Зависимость формы хроматографических пиков диэтилового эфира от температуры.
Объем пробы 5 мкл; 1 - 40С, 2 - 50С, 3 - 90С.
Подобный ход кривых легко объясняется с точки зрения дипольных моментов молекул (диэтиловый эфир 1,18*1030 Кл*м, метилэтилкетон 2,96 *1030 Кл*м). Адсорбция молекул имеющих более высокий дипольный момент должна сильнее зависеть от изменения энергии центров адсорбции с температурой. Но более высокое значение теплоты адсорбции для диэтилового эфира не объяснимо с данной позиции. Независимость удерживаемого объема и времени удержания диэтилового эфира от объема введенной пробы и независимость теплоты адсорбции от температуры указывает на постоянство энергии взаимодействия с поверхностью: возможно адсорбат мало чувствителен к изменению энергии центров адсорбции. Зависимость теплоты адсорбции метилэтилкетона от температуры указывает на более высокую чувствительность адсорбата к изменению энергии адсорбционных центров от температуры (активационная адсорбция [15]). Наблюдаемые зависимости объяснимы, если предположить, что диэтиловый эфир взаимодействует с поверхностью через d-центры, а диметилкетон и метилэтилкетон через поверхностные группы. Активность гидроксильных групп понижается с увеличением температуры, а d-центров увеличивается в связи с ослаблением экранирующего эффекта. Расчет числа поверхностных групп способных к адсорбции [18] кетонов дает величину порядка 1010-12м-2, что хорошо коррелирует с литературными данными для оксидов в растворе [19].
В качестве соединения способного адсорбироваться на оба типа центров нами был выбран пиридин рис. 8. -электронная система является донорно-акцепторным центром, а азот кислотно-основным.
Исследование адсорбции пиридина показывает очень специфическую зависимость от температуры. Для пиридина отмечается большой (до1,5 мкл) необратимо сорбирующий объем. Исследование зависимости отклика от времени между моментами ввода проб показывают, что отклик зависит от периода между вводом проб. Последовательный ввод по 0.2 мкл дает отклик после суммарного ввода 1.2 мкл за 3 минуты (при массе навески адсорбента 0.189 г). Но рассчитанное количество центров на единицу площади представляется нам сомнительной величиной (как указано выше). Из соотношения теплот адсорбции бензола (92 кДж*моль-1г-1) и пиридина (410 кДж*моль-1г-1) при температуре 353К нами рассчитаны вклады различных типов взаимодействий. Вклад взаимодействия кислотно-основных центров в 5 раз выше, чем донорно-акцепторных. Теплота адсорбции практически равна теплоте образования солей пиридиния. Этот вывод подтверждает факт резкого уменьшения теплоты адсорбции с повышением температуры, связанный с уменьшением вклада поверхностных гидроксильных групп в адсорбционные взаимодействия, показанный на зависимость адсорбции углекислого газа от температуры рис.11.

Рис.11. Зависимость времени удержания СО2 от температуры.
С возрастанием температуры увеличивается теплота адсорбции углекислого газа достигая максимума при температуре близкой к 100С. Перестройка координации поверхностных групп при более высокой температуре приводит к уменьшению количества адсорбированного углекислого газа. Обращение адсорбционного равновесия связано с образованием высококоординированных гидроксильных групп [16]. Это приводит к увеличению энергии необходимой для образования поверхностных комплексов [5, 18] и уменьшает вклад поверхностных групп в адсорбционное взаимодействие с повышением температуры.
Выводы
1. Показано, что на поверхности -FeOOH, вклад ионов железа в адсорбционные взаимодействия много ниже чем поверхностных гидроксильных групп (поверхностные группы экранируют d-центры).
2. В -FeOOH имеются поры сравнимые с размерами молекул гептана (объем которых может быть оценен).
3. В отличие от бензола и метилэтилкетона диэтиловый эфир имеет аномально высокую теплоту адсорбции
4. Адсорбционные свойства поверхностных гидроксильных групп сильно зависят от температуры.
Литература
- Шведт Г. Хроматографические методы в неорганическом анализе: Пер. с англ.-М, 1984.-252с.
- Молекулярная теория адсорбции газов на неспецифических адсорбентах. Пошкус Д.П. М., «Наука»,1970, стр. 483.
- Основы аналитической химии. В 2 кн. Кн.1. Ю.А.Золотов,Е.Н.Дорохова,В.И.Фадеева и др.М.: Высш. шк.,2002.351с.
- Количественный анализ хроматографическими методами. Под ред. Э.Кэц: Пер с англ. М.:Мир,1990.-320с.
- Давыдов А.А. ИК-спектроскопия в химии поверхности окислов. Новосибирск: Наука, 1984. с.246.
- Киселев А.В., Лыгин В. И. Инфракрасные спектры поверхностных соединений и адсорбированных веществ. М., 1972. 459 с., 15
- Фридрихсберг Д. А. Курс коллоидной химии. Учебник для вузов.- Л.: Химия, 1984.
- Oxides and Oxide Filme. Ed. Diggle J.W. Marcel Dekker: N. Y. 1972. V. 1. P. 319-517;1973. V. 2. Р. 231-387.
- Нечаев Е.А. Хемосорбция органических веществ на оксидах и металлах. Харьков: Высш.шк., 1989. С. 38.
- Руководство по неорганическому синтезу: В 6-ти т. Т.5:пер. с нем. / Под ред. Г. Брауэра. М.: Мир, 1985. C.1752.
- Кузнецов С.В., Батраков В.В., Хлупов А.Ю. К вопросу о синтезе окси-гидрокси форм железа./ Научные труды Московского педагогического государственного университета. Серия: естественные науки. - М.: Прометей, 2000. с. 256-263.
- Органическая электрохимия под ред. В.А. Петросяна и Л.Г. Феоктистова Т.2 «Москва. Химия. 1988г.470-1024с.
- Дамаскин Б.Б., Петрий О.А., Подловченко Б.И. и др.; Под ред. Дамаскина Б.Б.. Практикум по электрохимии. М.: Высш. Шк., 1991. С. 23-27.
- Практикум по химии поверхностных явлений и адсорбции» Б.В.Айвазов.М., «Высш. Школа»,1973,208 с .
- Газохроматографическое исследование адсорбционных свойств очищенного диализом -Fe2O3/Л.М. Воробьева, О.Г. Ларионов, А.Е. Чалых// Ж Ф Х, 1992. –T. 66, N11.-C. 3045-3051.
- Исследование свойств -Fe2O3 газохроматографическим методом /Е.В. Загоревская, Н.В. Ковалева, Н.С. Куликов, Ю.С. Никитин, И.С. Протонина, Б.В. Щербинин// Ж Ф Х, 1989. –T. LXIII, N12.-C. 3289-3296.
- Исследование адсорбции салициловой кислоты на -FeOOH/ С.В. Кузнецов, О.М. Шиш//Материалы рег. научно-технической конференции «Вклад ученых и специалистов в национальную экономику».Брянск: 20-21 мая 2004.-Т2.-С. 59-60.
- Электронные явления в адсорбции и катализе на полупроводниках. Лекции на международном симпозиуме. М.:1968.с.399.
- Горичев И.Г., Изотов А.Д., Кишкина Н.А., Кузнецов А.В. и др. Использование представлений о строении двойного электрического слоя в методах экспериментального определения и расчета констант кислотно-основных равновесий на границе оксид/электролит. М.: Российский университет дружбы народов. 2002г, с. 86.