
Тонкоструктурные спектры и электронно-колебательные взаимодействия сопряженных молекул цепочечного строения
| Вид материала | Автореферат |
- Реферат Отчет, 51.81kb.
- Исследование процессов взаимодействия молекул водорастворимого фуллерена с магнитными, 37.88kb.
- Программа дисциплины дпп. Ф. 02 Строение молекул и основы квантовой химии, 160.77kb.
- Молекулярная структура вещества. Скорости газовых молекул, 140.08kb.
- Физические величины, измеряемые в аэрогидромеханике и теплофизическом эксперименте., 39.57kb.
- 1. Некоторые вопросы строения веществ, 1501.26kb.
- Программа Государственного экзамена по подготовке магистра по направлению «Радиофизика», 49.35kb.
- Курс лекций «Основы радиоэлектроники» Часть Сигнал и его свойства. Линии передачи, 33.21kb.
- I. Основы физических процессов в ядерных реакторах, 559.27kb.
- Стереохимия, область химии, изучающая пространственное строение молекул и влияние этого, 19.63kb.
На правах рукописи
Васильева Ирина Александровна
ТОНКОСТРУКТУРНЫЕ СПЕКТРЫ
И ЭЛЕКТРОННО-КОЛЕБАТЕЛЬНЫЕ ВЗАИМОДЕЙСТВИЯ СОПРЯЖЕННЫХ МОЛЕКУЛ ЦЕПОЧЕЧНОГО СТРОЕНИЯ
Специальность 01.04.05 - Оптика
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
доктора физико-математических наук
Москва – 2009
Работа выполнена в Проблемной научно-исследовательской лаборатории спектроскопии сложных органических соединений и на кафедре общей и экспериментальной физики факультета физики и информационных технологий Московского педагогического государственного университета
Научный консультант: доктор физико-математических наук
профессор, Гольцман Григорий Наумович
Официальные оппоненты: доктор физико-математических наук
профессор, Витухновский Алексей Григорьевич
доктор физико-математических наук
профессор, Коротаев Олег Николаевич
доктор физико-математических наук
профессор, Майер Георгий Владимирович
Ведущая организация: ФГУП Научно-исследовательский физико-химический институт им. Л.Я. Карпова, Москва.
Защита состоится 1 марта 2010 года в 16.00 на заседании диссертационного совета
Д 212. 154.22 при Московском педагогическом государственном университете по адресу: 119435, г. Москва, ул. Малая Пироговская, д. 29, ауд. 30.
С диссертацией можно ознакомиться в библиотеке Московского педагогического государственного университета по адресу: 119992, г. Москва, ул. Малая Пироговская, д.1.
Автореферат разослан «___»_________2009 года
Ученый секретарь
диссертационного совета Ильин В.А.
Спектральные исследования являются одним из главных источников информации о строении молекул, их оптических свойствах и взаимодействии с окружающей средой.
Вторая половина ХХ века ознаменовалась бурным развитием фотофизики многоатомных органических молекул сложного строения. Этому в значительной степени способствовало интенсивное развитие в тот же период, методов тонкоструктурной электронно-колебательной спектроскопии, которые позволили раскрыть особенности электровозбужденных состояний таких сложных систем и механизмы их дезактивации. В число указанных методов входят: метод Шпольского [1,2], метод селективной спектроскопии [3], метод сверхзвуковой охлажденной струи [4] и развивающийся в настоящее время, метод спектроскопии одиночных молекул [5,6]. С их помощью были изучены тонкоструктурные спектры флуоресценции, фосфоресценции и поглощения огромного числа сложных соединений (полициклические ароматические углеводороды – ПАУ, N-гетероароматические соединения, полициклохиноны, диоксины, порфирины и т.д.).
Для углубления понимания процессов взаимодействия в системе примесный центр - кристаллическое окружение, когда примесью является молекула сложного органического соединения, важно знание закономерностей в формировании вибронных спектров. Тонкоструктурные спектры позволяют установить общие связи строения молекул и их электронных спектров, выявить закономерности в вибронных переходах и проявления внутримолекулярных взаимодействий [7,8].
Диссертационная работа посвящена получению тонкоструктурных спектров и выяснению природы нарушения зеркальной симметрии в распределении интенсивностей в сопряженных спектрах флуоресценции и возбуждения флуоресценции органических люминофоров цепочечного строения в н-парафиновых матрицах Шпольского. Наиболее результативен для решения подобных задач одновременный сравнительный анализ сопряженных спектров флуоресценции и поглощения (возбуждения флуоресценции), применяемый в данной работе, позволяющий количественно анализировать распределение интенсивностей по спектрам.
Молекулы большинства ранее исследованных соединений обладают «жесткой» структурой, например полициклические ароматические углеводороды (ПАУ). Соединения, имеющие цепочечную структуру, изучены гораздо меньше. Такие молекулы обладают возможностью изменения своей топологии и могут существовать в растворах в виде нескольких конформеров (стереоизомеров). Спектрально-люминесцентные свойства многих люминофоров цепочечного строения при комнатной температуре в растворителях с различной полярностью достаточно хорошо изучены [см., например, 9,10]. В то же время попытки исследовать такие соединения методами тонкоструктурной спектроскопии долгое время оставались малоудачными. Спектры многих из этих соединений пробовали получать методом Шпольского при низких температурах. Однако при 77 К удавалось регистрировать спектры только с незначительным разрешением колебательной структуры по сравнению со структурой спектров при комнатной температуре. При понижении температуры до 4,2 К узкие полосы в спектрах молекул цепочечного строения накладываются на интенсивный сплошной фон. Вибрационный анализ и количественная оценка параметров внутримолекулярных взаимодействий по таким спектрам проводится с большой погрешностью, так как невозможно оценить относительную интенсивность вибронного перехода.
Актуальность исследования определяется тем, что цепочечные молекулы представляют собой основную структурную единицу громадного класса веществ – природных и синтетических полимеров. В конце прошлого столетия было сделано важнейшее открытие, удостоенное Нобелевской премии по химии за 2000 г. Алан Хигер, Алан Мак-Диармид и Хидеки Сиракава получили эту премию за исследования квазиметаллической электропроводности органических сопряженных полимеров, допированных окислителями или восстановителями. Одной из особенностей линейных -электронных молекул как полупроводниковых систем со сравнительно небольшой запрещенной зоной ( 3 эВ) является интенсивное поглощение света в видимой области спектра. На этом основано использование подобных соединений в материалах для преобразования световой энергии в электрическую. В этих целях широко применяются замещенные полиены и стильбены, донорно-акцепторные молекулы и мероцианины, а также и другие линейные молекулы с -сопряжением. Подобные соединения используются для создания новых перспективных материалов для молекулярной электроники и нелинейной оптики. Замещенные арилполиены нашли применение в бессеребряной фотографии, сцинтилляционной технике, в лазерах на органических красителях, в люминесцентной дефектоскопии и ряде других отраслей науки и техники. Не будет преувеличением сказать, что все живые организмы построены в основном из цепочечных молекул, например, белки, нуклеиновые кислоты, целлюлоза и ряд других веществ биологического происхождения. Интерес к этим соединениям резко возрос в начале 60-х годов прошлого века, когда стала понятна роль возбужденных электронных состояний полиеновых соединений (ретинолов, ретиналей и др.) в ряде важных фотобиологических процессов, в том числе и в процессе зрения. Полиеновые соединения, входящие в состав зрительных пигментов, обладают большой поглощательной способностью и довольно низкой вероятностью излучения. Для понимания механизма зрения на молекулярном уровне необходима информация о процессах, сопровождающих в этих молекулах поглощение квантов света. Отсутствие флуоресценции у полиеновых соединений, входящих в состав зрительных пигментов, затрудняет исследование их спектральных свойств. Поэтому использование флуоресцирующих линейных полиенов, в частности дифенилполиенов, как модельных соединений дает возможность получить более полную информацию о процессах, протекающих в зрительных пигментах. Линейные сопряженные соединения, из-за простоты их химического строения и легкости варьирования молекулярной топологии, традиционно служили «полигоном» для разработки новых квантовохимических моделей (см., например, [11-13]).
Для дальнейшего развития теории вибронных спектров сложных органических молекул необходимы новые данные по тонкоструктурным спектрам, тем более, впервые синтезированных соединений мало изученного класса органических соединений цепочечной структуры. Заметим, что в свое время толчком для разработки теоретического подхода к рассмотрению этой проблемы явилось открытие эффекта Шпольского. И в последующем, каждому существенному шагу в разработке упомянутой теории предшествовало накопление систематических экспериментальных данных.
Цель данного исследования – детальное изучение спектральных свойств массива впервые синтезированных линейных молекул различной химической структуры; получение информации для решения фундаментальной проблемы выяснения закономерностей в механизмах формирования вибронных спектров линейных органических люминофоров.
Для этого были применены экспериментальные методы исследования спектров в широком диапазоне температур (от 4.2 до 293 К), а также теоретический анализ спектров, позволяющий количественно определять вклад электронно-колебательных взаимодействий в формирование вибронных спектров органических соединений цепочечной структуры.
Одна из основных задач данной работы – выявить детальные механизмы вибронных переходов S0 S1 цепочечных –сопряженных молекул.
Для достижения этого необходимо было:
- Получить сопряженные спектры флуоресценции и поглощения (возбуждения флуоресценции) исследуемых соединений с развитой вибронной структурой.
- Выявить связь особенностей спектров со структурой молекул.
- Разработать метод моделирования экспериментально полученных спектров набором вибронных полос, состоящих из бесфононной линии и фононного крыла для определения относительной интегральной интенсивности каждого вибронного перехода.
- Проанализировать полученные спектры согласно современным теориям и оценить параметры внутримолекулярных взаимодействий.
- Проанализировать применимость адиабатической модели для анализа спектров линейных молекул, обладающих –сопряжением.
Объекты и методы исследований. Из всего многообразия линейных сопряженные систем, были выбраны молекулы, которые по своему строению и химическим свойствам представляют три группы соединений. Все эти соединения флуоресцируют. В первой группе исследовались ароматические соединения линейной структуры с общей формулой С6Н5 – (СН=СН)n – C6H5 (n=1, 2, 3, 4) и близкие к ним по строению; во второй группе – замещенные и производные от соединений первой группы: ,-дизамещенные полиены с n=2, 3, содержащие электронно-донорные заместители типа NH2, и N(CH3)2 и (или) электронно-акцепторные NO2, CN, а также кросс-сопряженные кетоны, в сопряженной системе которых есть мостик
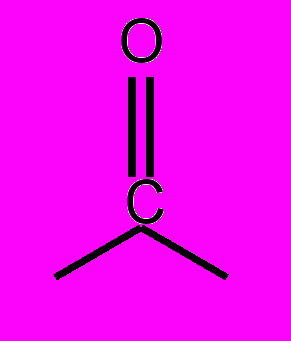 ; в третьей — гетероциклические соединения цепочечной структуры с общей формулой Z-Y-X-Y-Z, где X – фенильный или фурановый цикл, Y – оксазольный или оксадиазольный цикл и Z – фенильный или мезитильный цикл.
; в третьей — гетероциклические соединения цепочечной структуры с общей формулой Z-Y-X-Y-Z, где X – фенильный или фурановый цикл, Y – оксазольный или оксадиазольный цикл и Z – фенильный или мезитильный цикл. Сопряженные тонкоструктурные спектры флуоресценции и возбуждения флуоресценции были получены при 4,2 К в матрицах Шпольского. Для соединений, не растворившихся в н-парафинах, сопряженные спектры были получены в растворителях с различной полярностью при 77 К.
Кинетика затухания флуоресценции исследовалась методом однофотонного счета, позволявшим получать кривые затухания флуоресценции в наносекундной области при очень малых интенсивностях света и с достаточно хорошим временным разрешением (вплоть до 0.2 нс).
Научная новизна и значимость полученных результатов.
1) Впервые проведено систематическое исследование спектрально-люминесцентных свойств 3-х групп соединений цепочечной структуры. Всего было исследовано 41 соединение, 30 из которых были синтезированы впервые.
2) Определены закономерности в строении вибронных спектров линейных молекул в зависимости от их атомной структуры.
3) Разработан и применен на практике метод моделирования спектров, позволяющий по полученным спектрам определять относительную интенсивность вибронных переходов. Рассчитаны параметры франк-кондоновского (FC-) и герцберг-теллеровского (НТ-) взаимодействий для нормальных колебаний молекул исследуемых соединений.
4) Впервые дана количественная оценка параметров электронно-колебательного взаимодействия в -сопряженных молекулах и объяснена природа незеркальности спектров флуоресценции и возбуждения флуоресценции исследуемых соединений.
5) Сильная незеркальность сопряженных спектров флуоресценции и возбуждения флуоресценции качественно объяснена в рамках теории двухямных адиабатических потенциалов.
На защиту выносятся следующие положения
1. Данные о спектрально-люминесцентных свойствах 41 соединения класса линейных -сопряженных молекул, более 30 из которых синтезированы впервые: условия получения и регистрация тонкоструктурных спектров, результаты кинетических измерений длительности флуоресценции.
2. Метод, позволяющий оценить с определенной точностью относительную интенсивность каждого вибронного перехода в спектрах со слабо выраженной тонкой структурой, проявляющейся на интенсивном фоне. Суть метода в моделировании спектров набором вибронных полос, каждая из которых состоит из узкой бесфононной линии (БФЛ), описываемой функцией Лоренца, и широкого фононного крыла (ФК), описываемого функцией Гаусса. В процессе моделирования спектр получается практически идентичен экспериментальному.
3. Результаты вибрационного анализа тонкоструктурных спектров, полученных в процессе моделирования и расчета параметров внутримолекулярных взаимодействий. Доказательство существенной роли в формировании вибронных спектров рассматриваемого типа соединений НТ-взаимодействия, которое не только усложняет вибронную структуру спектра, но и влияет на интенсивности внутримолекулярных переходов.
4. Результаты анализа влияния на спектральные свойства -сопряженных соединений цепочечной структуры замещения одного атома водорода на функциональные группы, содержащие гетероатомы с разнотипными донорно-акцепторыми свойствами.
Практическая ценность работы заключается в том, что экспериментально установленные закономерности спектрально-люминецентных свойств вновь синтезированных соединений цепочечной структуры значительно дополняют ранее известные физико-химические данные о молекулах этого типа и могут быть использованы для интерпретации и прогнозирования оптических свойств органических люминофоров. Полученные в работе результаты могут быть использованы при разработке высокочувствительного спектрального метода анализа на содержание в образцах органических соединений цепочечной структуры. Разработанный и примененный метод для анализа экспериментальных спектров Шпольского, в которых БФЛ проявляется на значительном фоне, может быть применен для других классов соединений. Полученные в работе данные о внутримолекулярных электронно-колебательных взаимодействиях, формирующих вибронные спектры молекул цепочечного строения, открывают путь к дальнейшему развитию теории внутримолекулярных взаимодействий.
Личный вклад автора. В диссертации изложены результаты работ, выполненных автором лично и в соавторстве с коллегами. Исследования, связанные с развитием научного направления тонкоструктурной спектроскопии соединений цепочечного строения с 1980 г. по 2002 г. велись в тесном сотрудничестве с А.Н. Никитиной. Все экспериментальные спектральные данные были получены самим автором или под его руководством. Автор осуществлял анализ и обобщение полученных данных, участвовал в разработке предложенного метода моделирования спектров, интерпретировал полученные результаты и проводил все необходимые расчеты. Кинетические измерения проводились в сотрудничестве с З.А. Чижиковой и М.Д. Галаниным (Физический институт РАН им. П.Н. Лебедева)
Публикации. По результатам проведенных исследований опубликованы 46 работ, в том числе 19 в ведущих рецензируемых отечественных и зарубежных научных журналах, а также в материалах Всесоюзных, Всероссийских и международных конференций. Список публикаций приведен в конце реферата.
Апробация результатов. Основные результаты выполненных исследований были доложены и обсуждены на IV Всесоюзном совещании по фотохимии (Ленинград, 1981 ); XIX, XX Всесоюзных съездах по спектроскопии (Томск, 1983; Киев, 1988); Всесоюзном совещании “Люминесценция молекул и кристаллов” (Таллинн, 1987); Всесоюзном семинаре “Спектроскопия свободных сложных молекул” (Минск, 1989); Всесоюзном совещания по молекулярной люминесценции (Караганда, 1989); VI Всесоюзной конференции “Органические люминофоры и их применение в народном хозяйстве” (Харьков, 1990); Всесоюзной конференции по люминесценции, посвященной 100–летию со дня рождения ак. С.И. Вавилова (Москва, 1991); XI, XII украинских школах–семинарах “Спектроскопiя молекул та кристаллiв” (Киев, 1993; 1995), международной научной конференции “Физика и химия органических люминофоров — 95”, (Харьков, 1995); XIII национальной школе–семинара с международным участием “Спектроскопiя молекул та кристалiв” (Сумы, 1997); XIV международной школе–семинаре “Spectroscopy of molecules and crystals” (Одесса, 1999); XXII и XXIII съездах по спектроскопии (Звенигород, Моск. обл., 2001 г. и 2005 г.) международной конференции по люминесценции, посвященной 110–летию со дня рождения академика С.И. Вавилова (Москва, 2001 г); международной конференции 14-th International Conference on Dynamical processes in excited states of solids (Christchurch, New Zealand, 2003); первой межрегиональной научно–практической конференции «Наука и молодежь в ХХI веке» (Троицк, Моск. обл., 2004); 4-й Всероссийской конференции «Молекулярное моделирование» (Москва, 2005 г.); XV Международный симпозиум по молекулярной спектроскопии высокого разрешения HighRus-2006 (Нижний Новгород, 2006г.); XVIII International School-Seminar “Spectroscopy of molecules and crystals”, Beregove, Crimear, Ukraine, 20.09 – 27.09.2007.
Структура и объем диссертационной работы. Диссертация состоит из введения, шести глав, основных результатов и выводов, приложения и списка цитируемой литературы. Диссертационная работа изложена на 263 страницах, включая 51 рисунок, 28 таблиц и список литературы из 318 наименований.
Содержание работы. Во введении кратко сформулирована цель диссертационной работы, обоснована ее
актуальность, защищаемые положения, научная новизна, выбор объектов и методов исследования.
В первой главе кратко рассмотрены основные положения современной теории электронно-колебательных спектров органических молекул в адиабатическом и гармоническом приближении [см. например, 7,8], даны аналитические выражения, описывающие форму вибронной полосы и распределение интенсивностей по спектрам флуоресценции и поглощения примесной молекулы в адиабатическом и гармоническом приближении в пренебрежении эффектом Душинского. Эти выражения позволяют анализировать экспериментальные спектры и количественно оценивать влияние внутримолекулярных взаимодействий на их формирование в случае относительно небольшой незеркальности между сопряженными спектрами, как по распределению интенсивностей, так и по частотам. В то же время сильная незеркальность между сопряженными спектрами может быть связана с наличием двух минимумов у адиабатического потенциала в основном и (или) возбужденном состояниях молекулы [14,15]. Анализ сопряженных спектров молекул цепочечной структуры в рамках представленной теории является важной и актуальной задачей.
Вторая глава посвящена объектам и методам исследования и применяемым в данной работе экспериментальным методикам. В работе исследованы 41 соединение с -сопряжением цепочечной структуры, причем 25 из них были синтезированы впервые в ИОХ РАН им Н.Д. Зелинского, Москва (Красной Ж.А. с сотрудниками, Беленьким Л.И. с сотрудниками) и 5 соединений — в Институте монокристаллов, Харьков (группой Афанасиади Л.М). Выбор объектов был обусловлен тем, что мы стремились изучить возможность получения тонкоструктурных спектров, а также выяснить общие признаки и особенности механизма формирования вибронной структуры для цепочечных молекул разного химического строения. Структурные формулы исследованных соединений представленные в таблице 1.
Все исследованные соединения можно по структурным особенностям подразделить на три группы: 1) ароматические углеводороды с виниленовыми группами: стильбен, дистирилбензол, дифенилполиены; 2) производные и замещенные этих углеводородов: а) содержащие электронно-донорные заместители (типа NH2, и N(CH3)2) и (или) электронно-акцепторные (NO2, CN); б) кросс-сопряженные производные этих соединений; 3) соединения цепочечной структуры с пятичленными гетероциклами.
Таблица 2.1. Структурные формулы молекул исследованных соединений
Первая группа
| п/п | Структурная формула | | № п/п | Структурная формула |
| 1 | 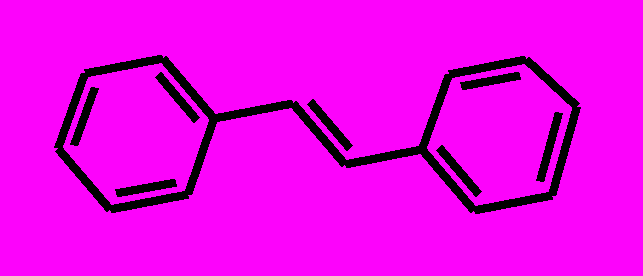 | 2 |  | |
| 3 |  | 4 |  | |
| 5 | 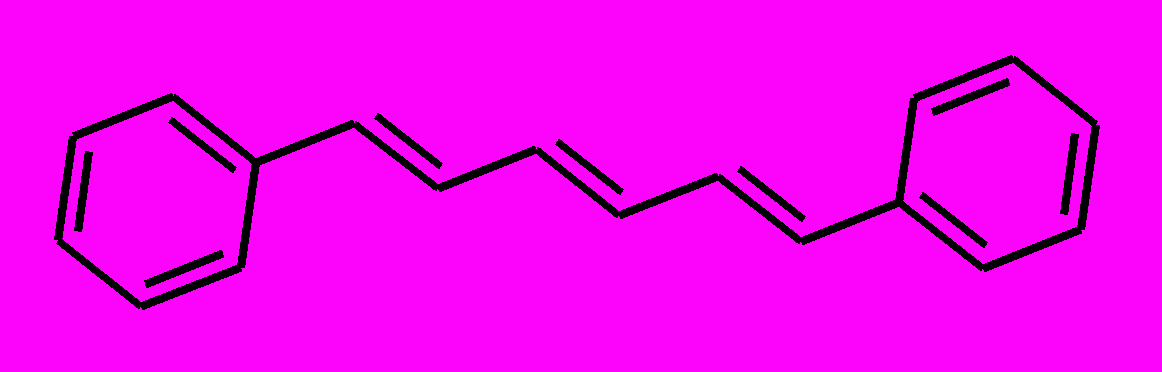 | 6 | 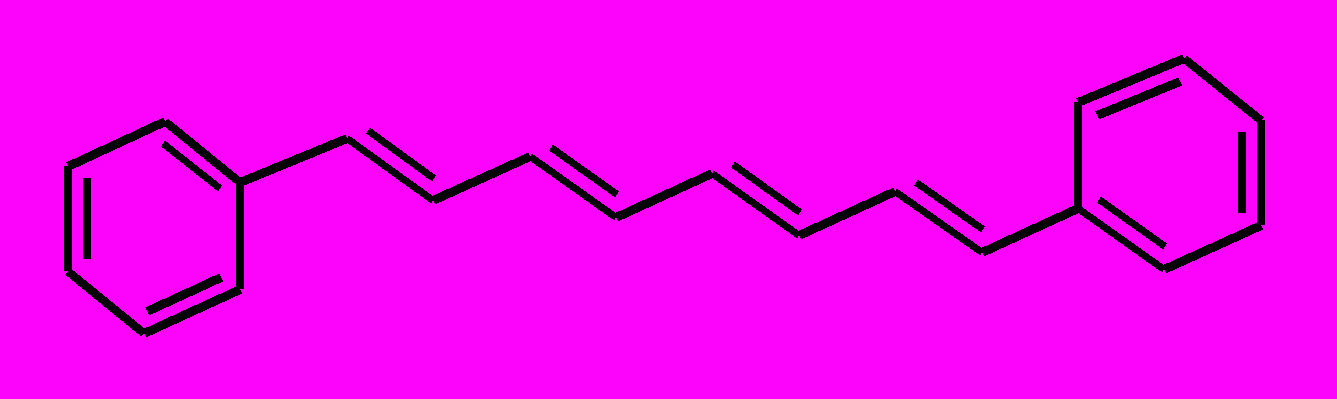 | |
| 7 | 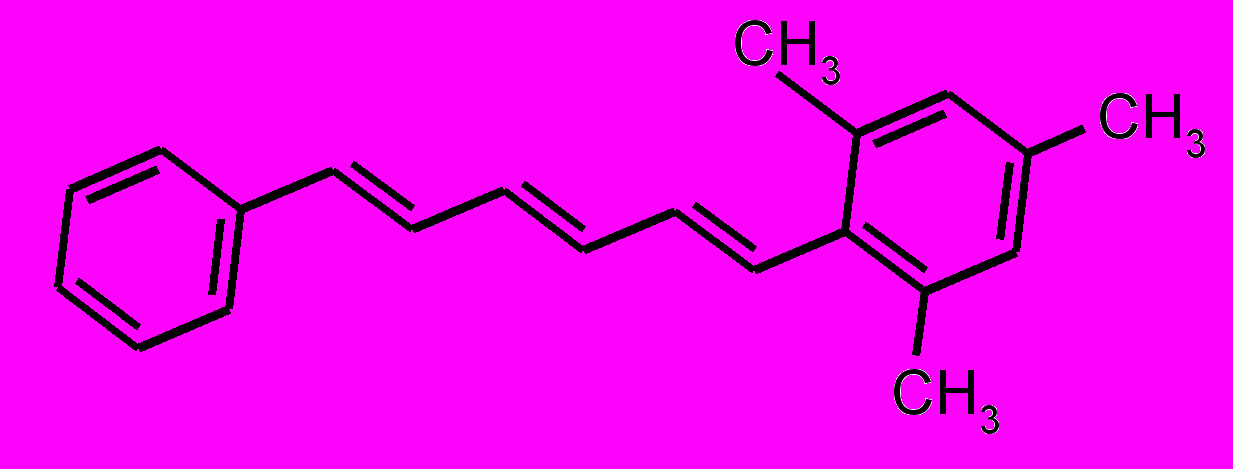 | | |
Вторая группа
| 8 | 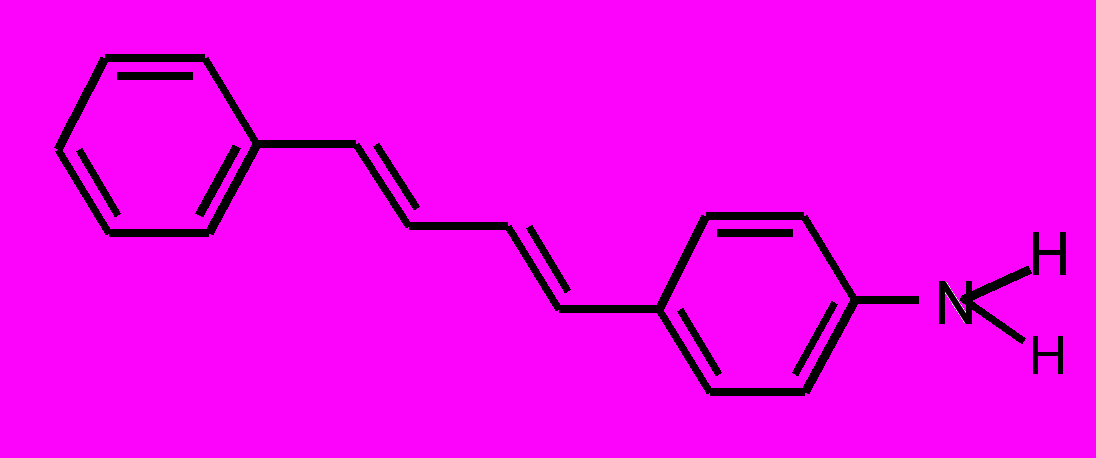 | | 9 | 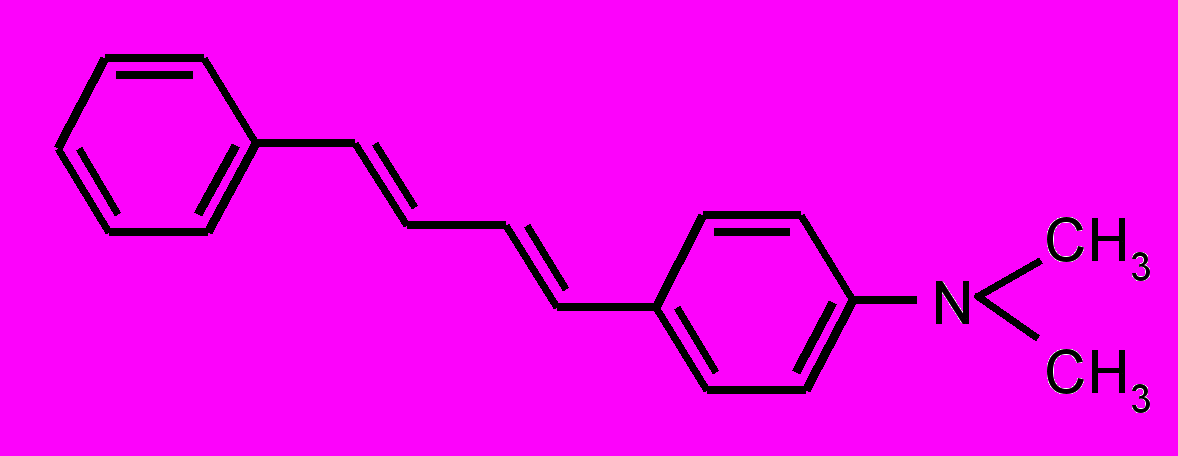 |
| 10 | 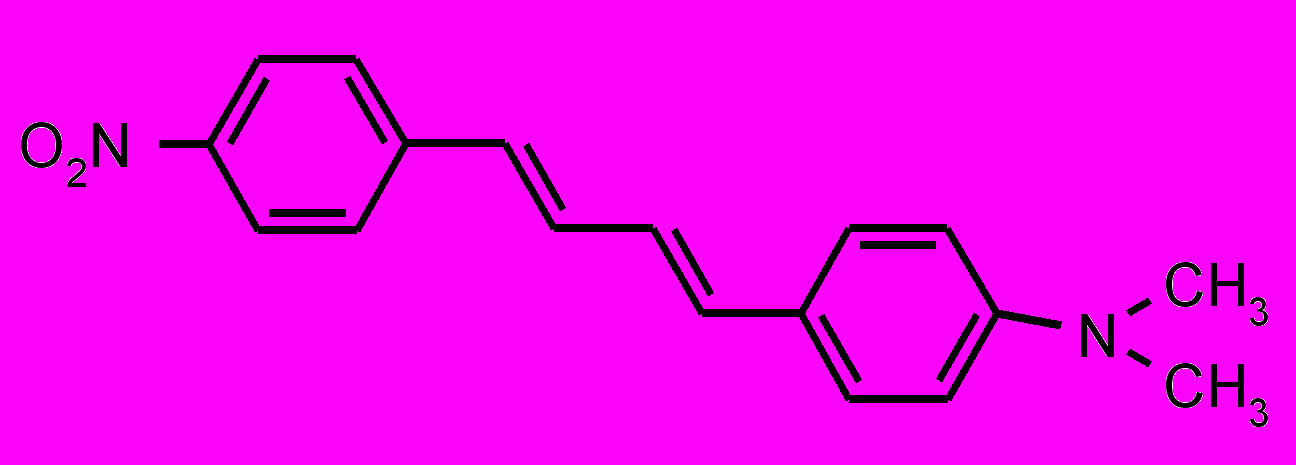 | | 11 | 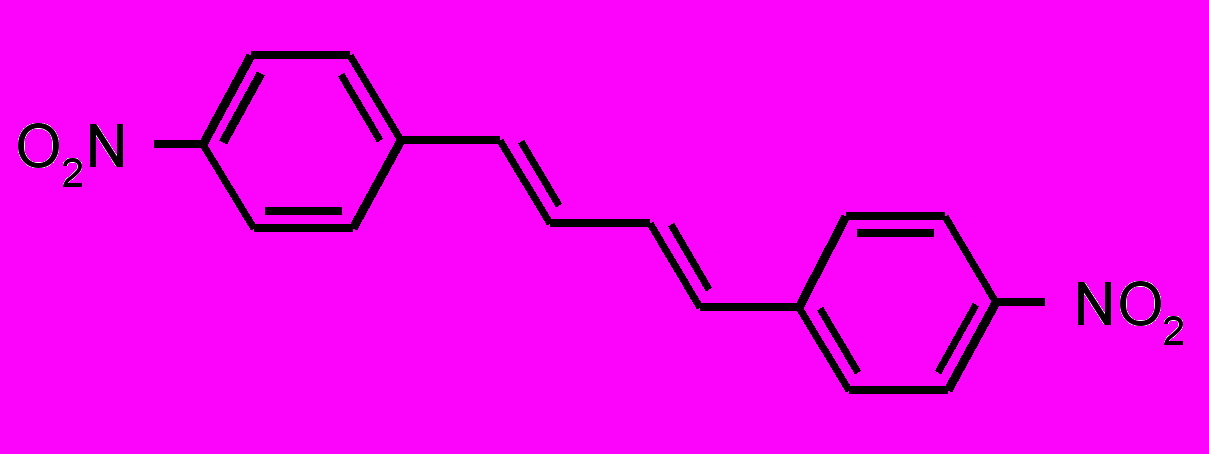 |
| 12 |  | 13 | 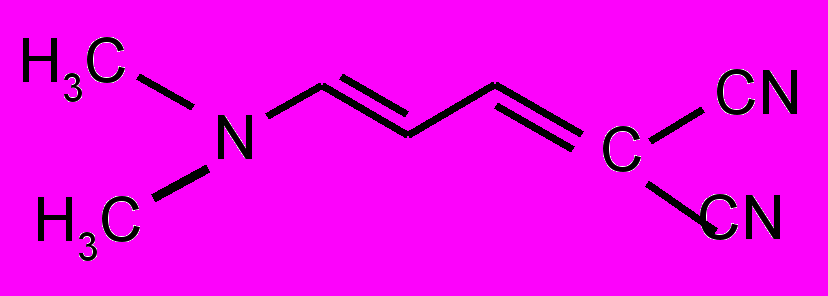 | |
| 14 | 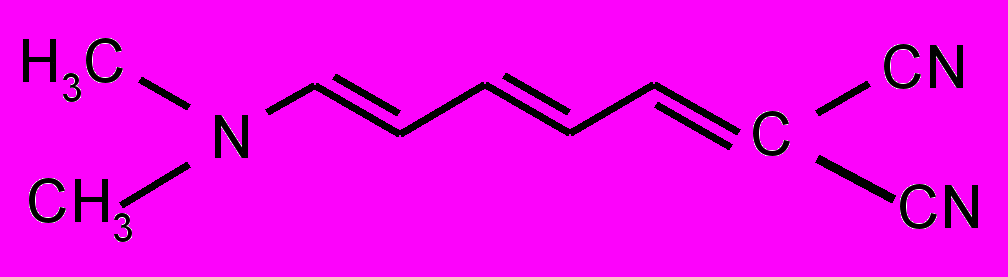 | | | |
| 15 | 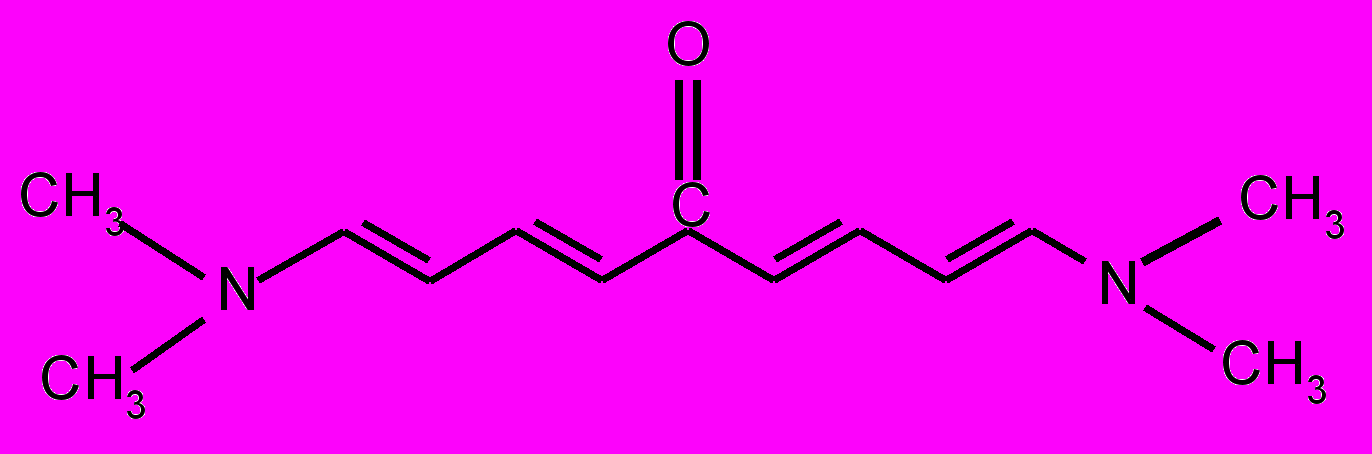 | 16 | 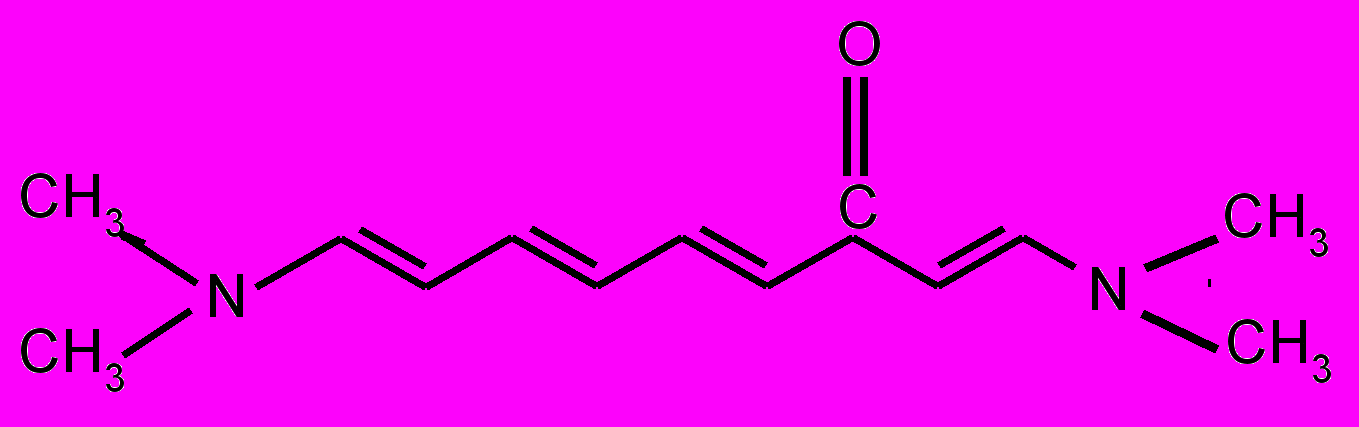 | |
| 17 | 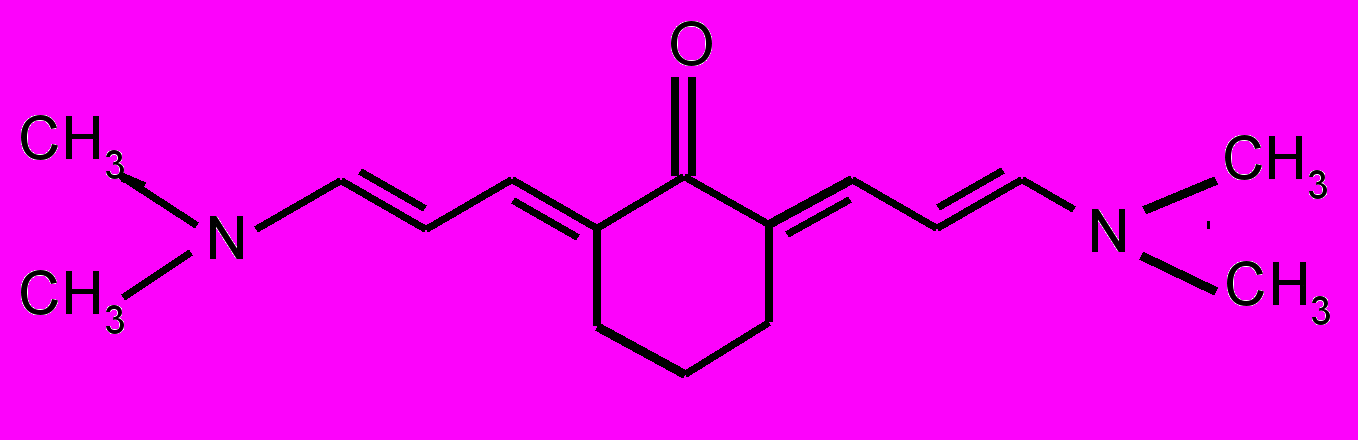 | 18 | 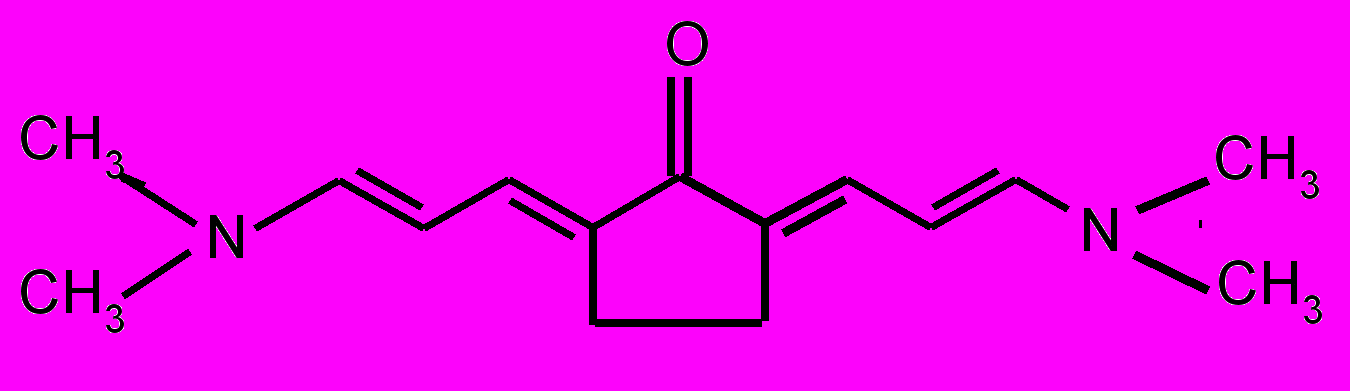 | |
| 19 | 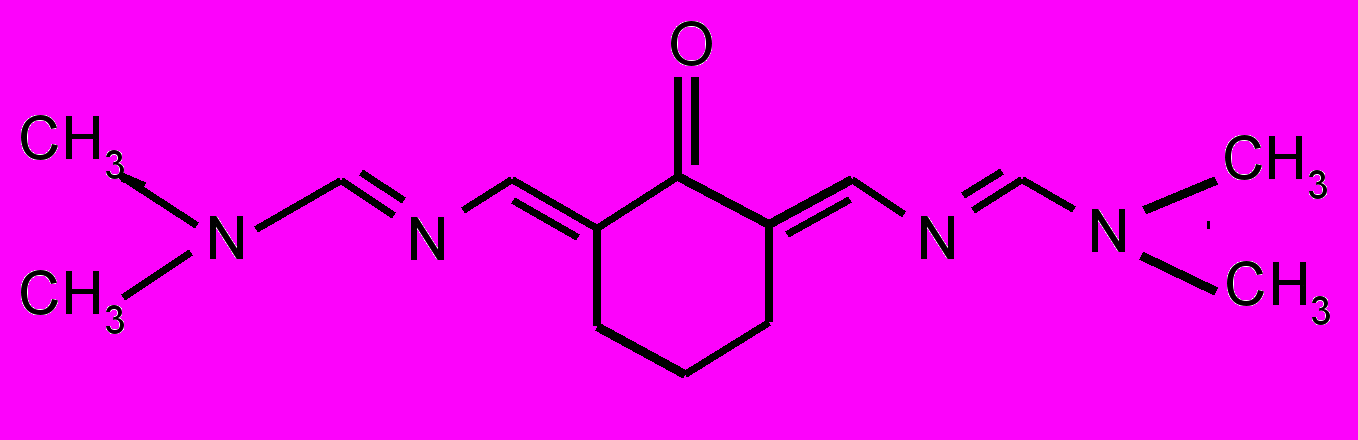 | 20 | 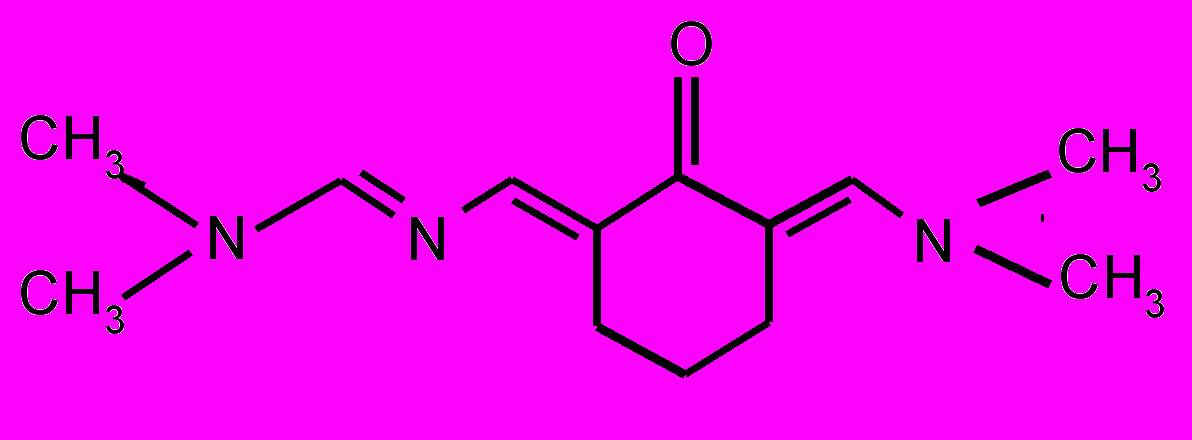 | |
| 21 | 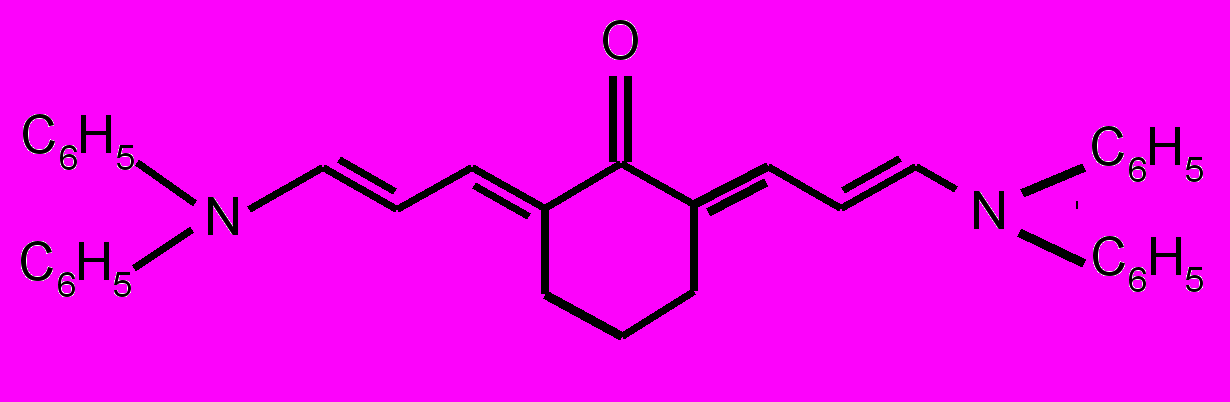 | 22 | 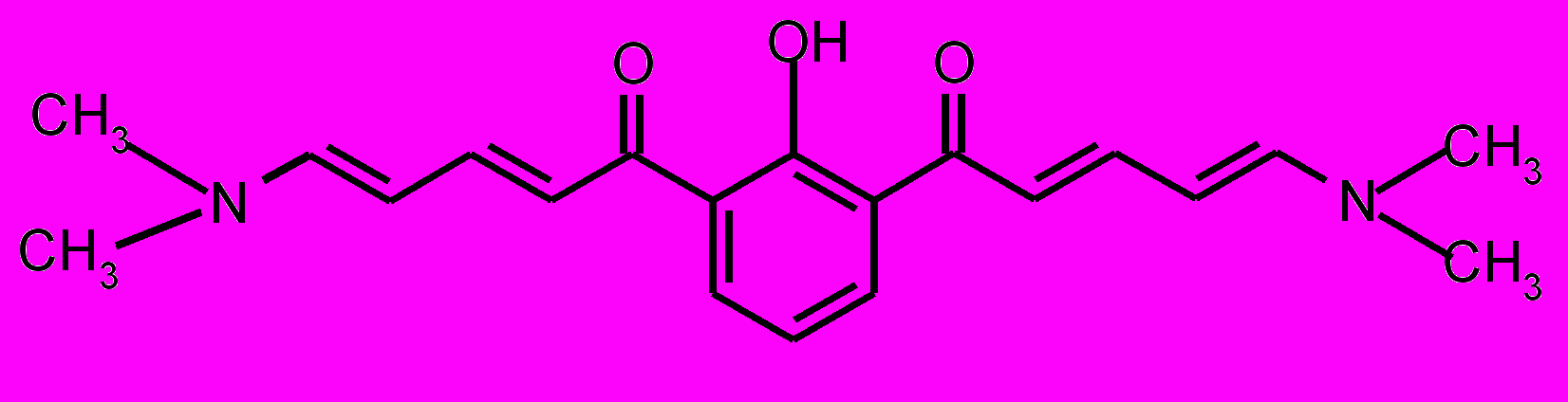 | |
| 23 | 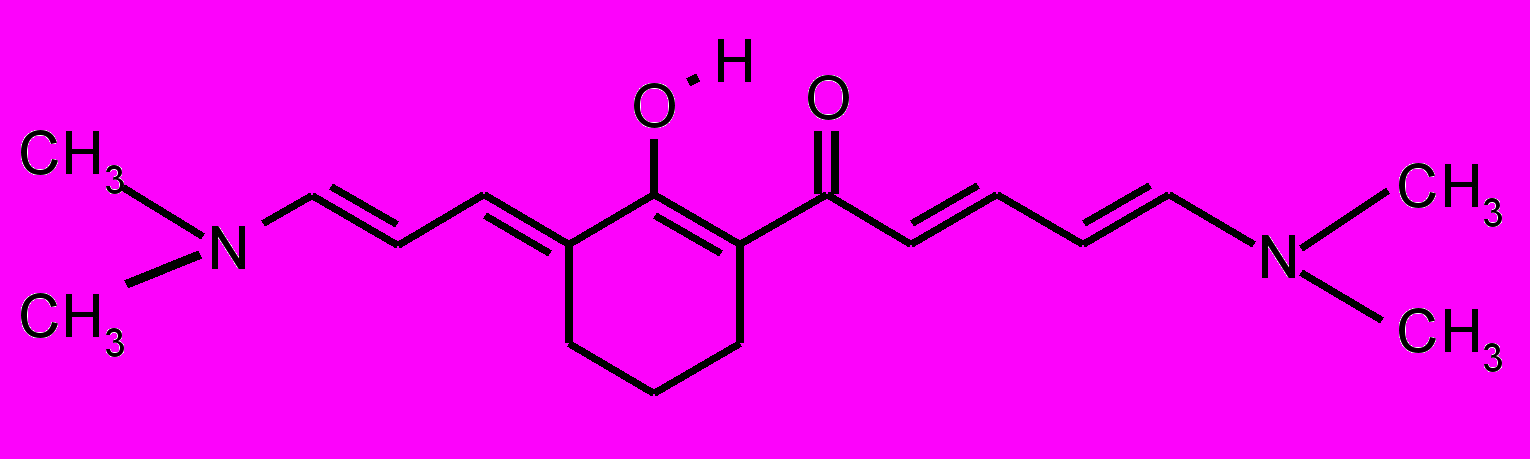 | 24 | 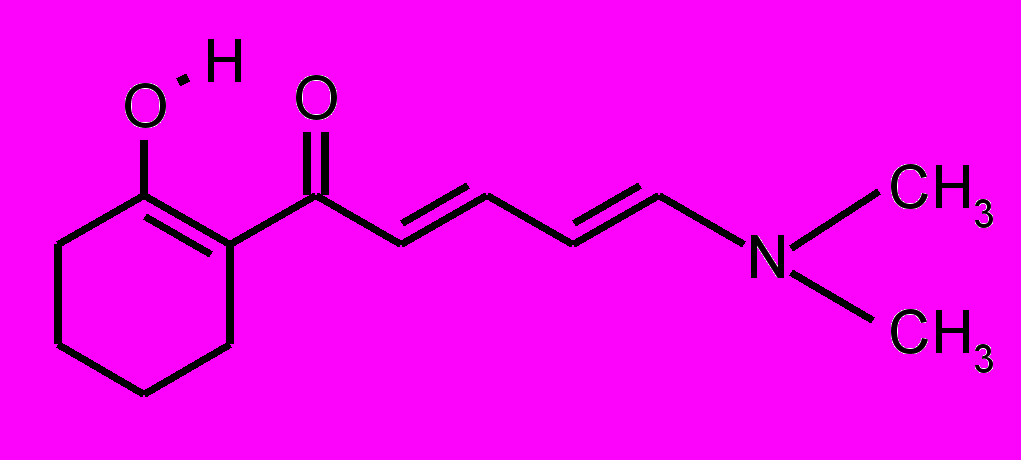 | |
| 25 | 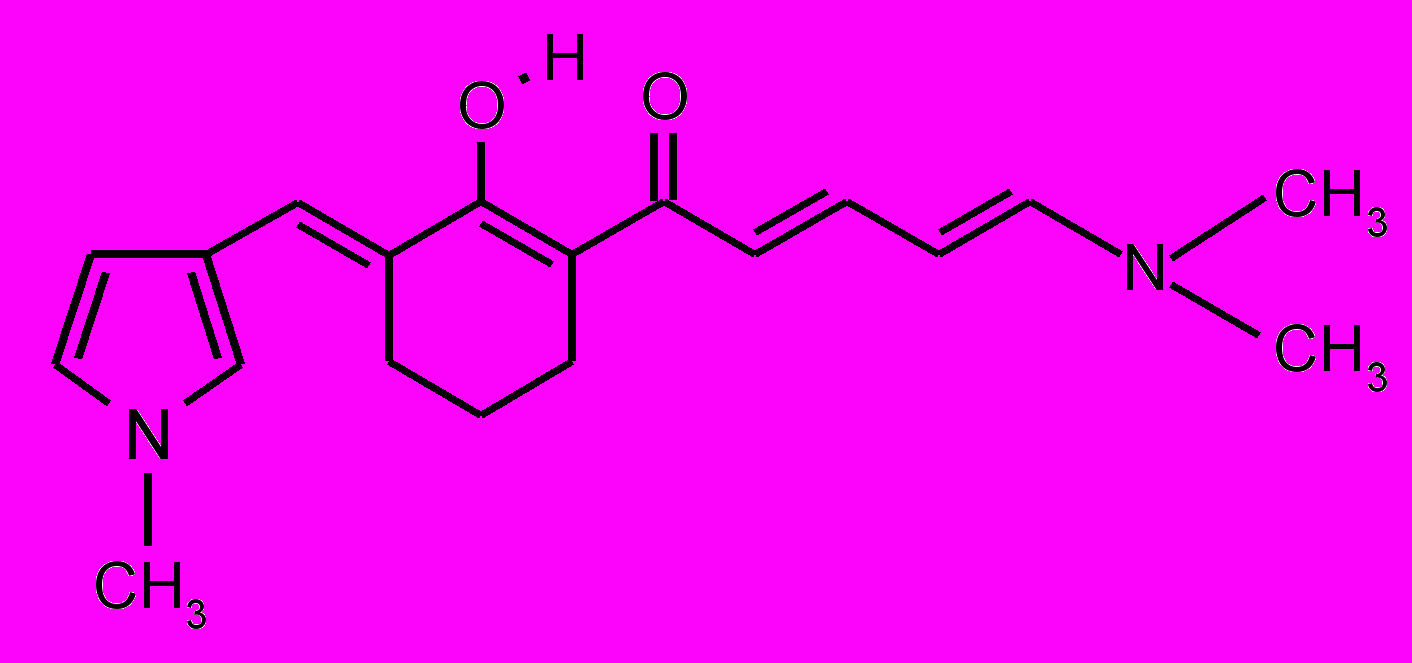 | 26 | 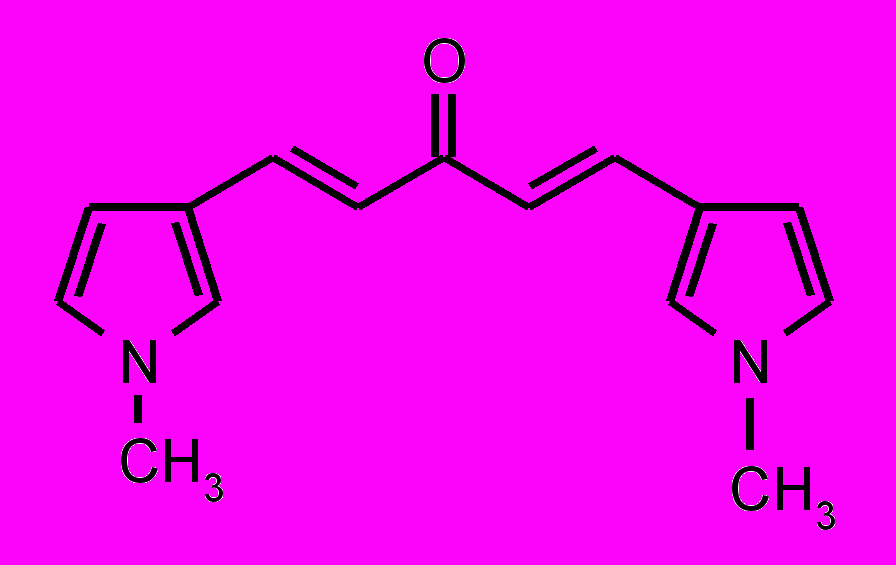 | |
| 27 | 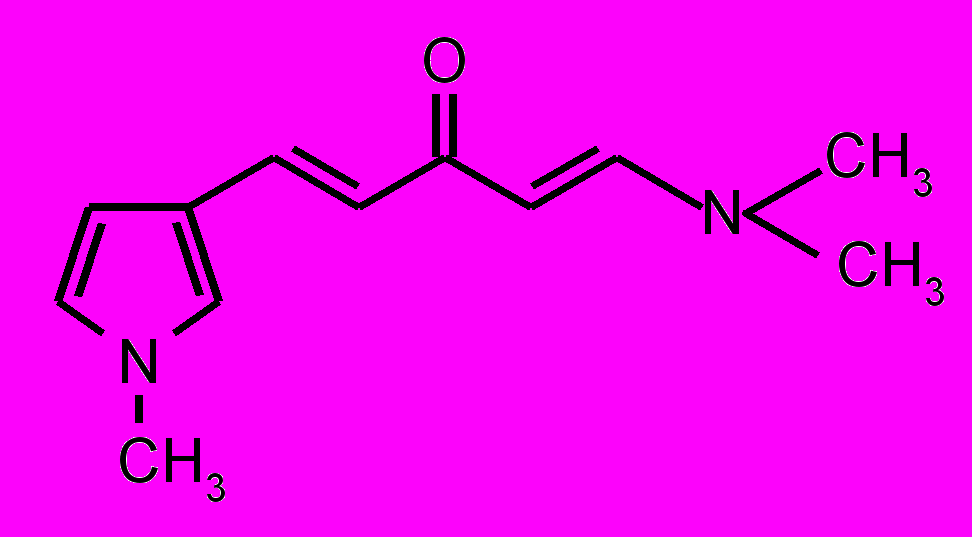 | 28 | 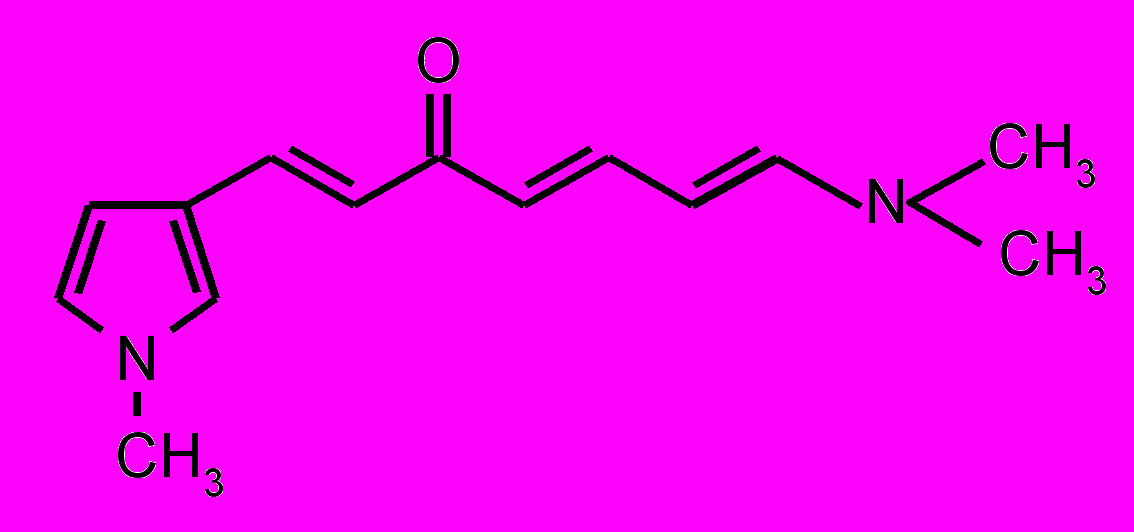 | |
| 29 | 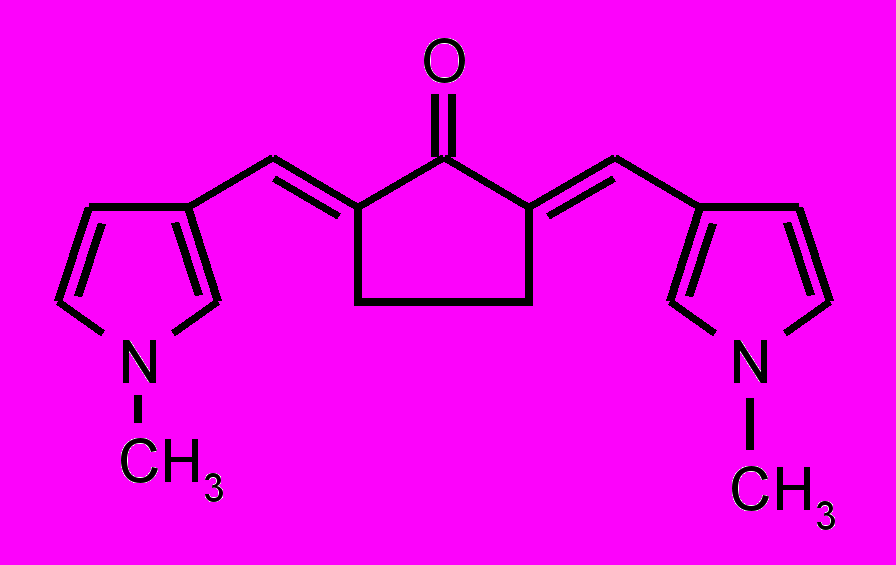 | 30 | 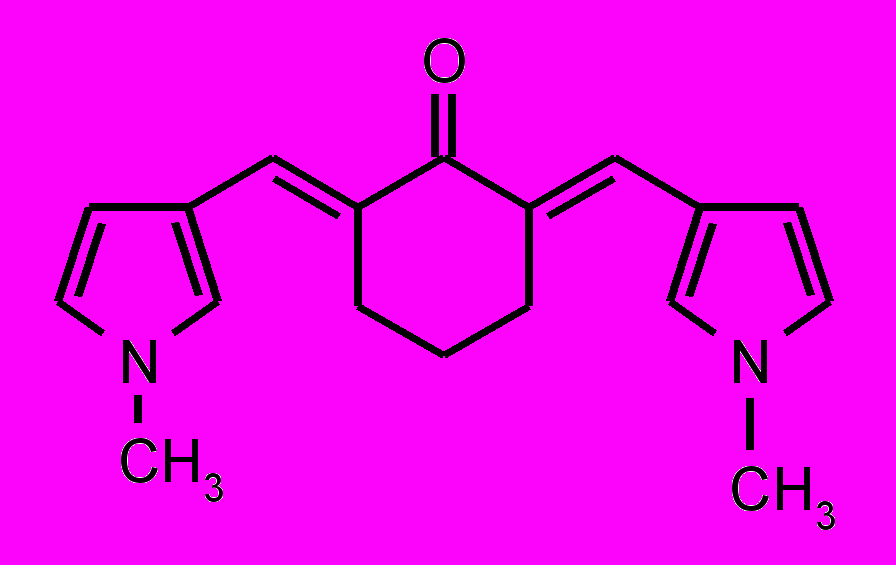 | |
| 31 | 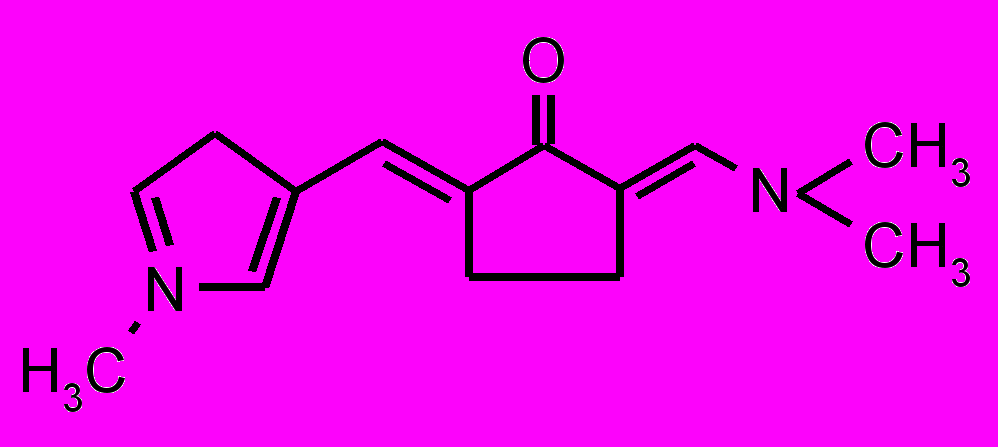 | 32 | 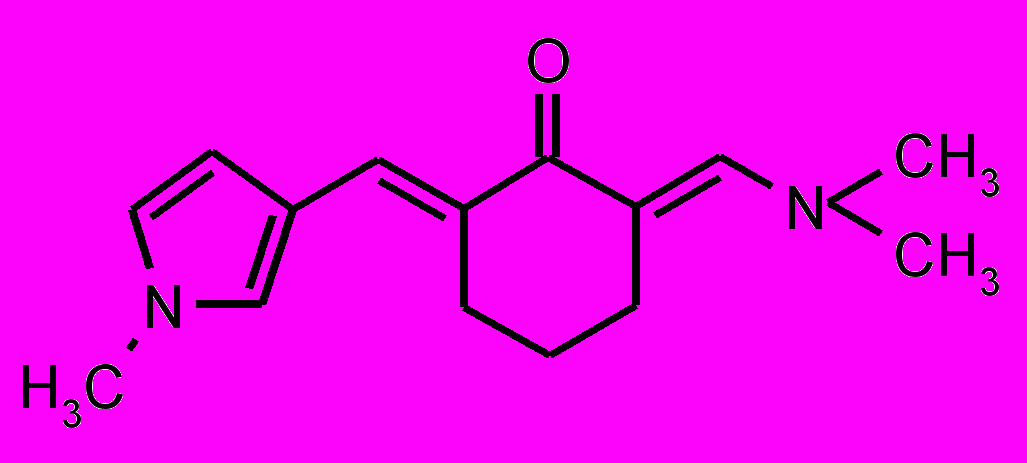 |
Третья группа
| 33 | 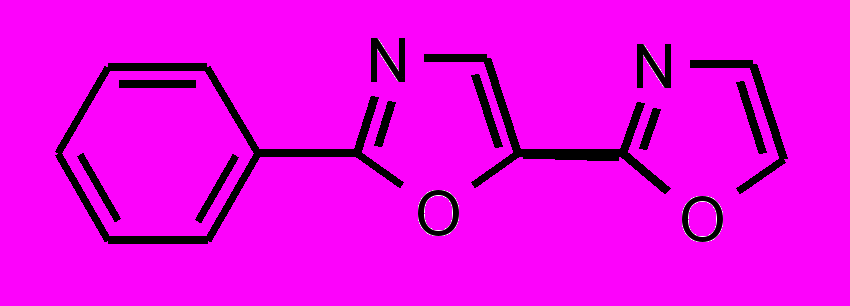 | | 34 | 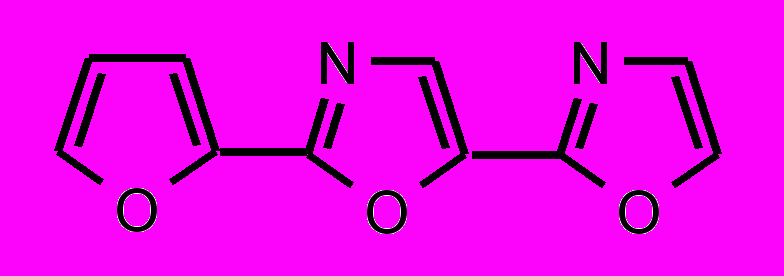 |
| 35 | 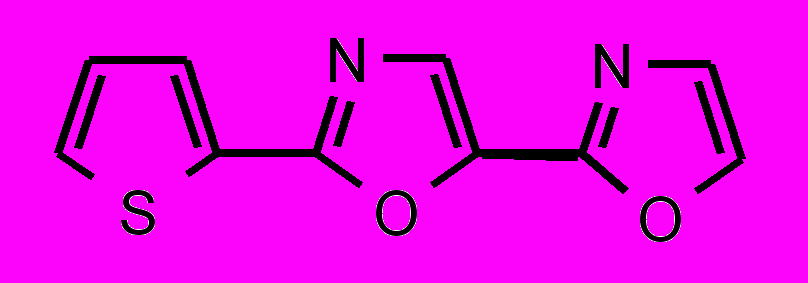 | 36 | 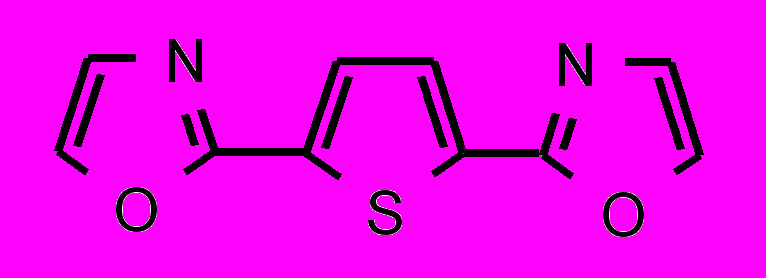 | |
| 37 | 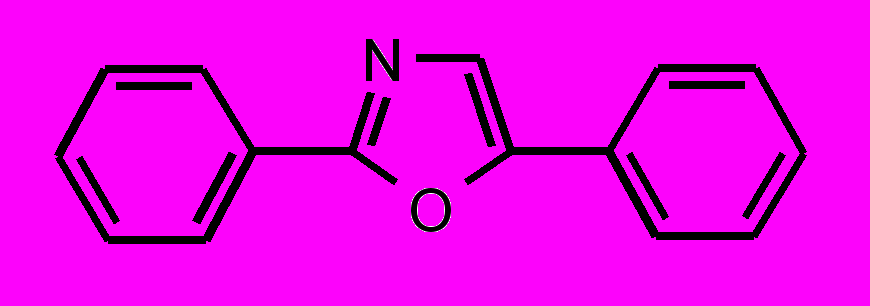 | 38 | 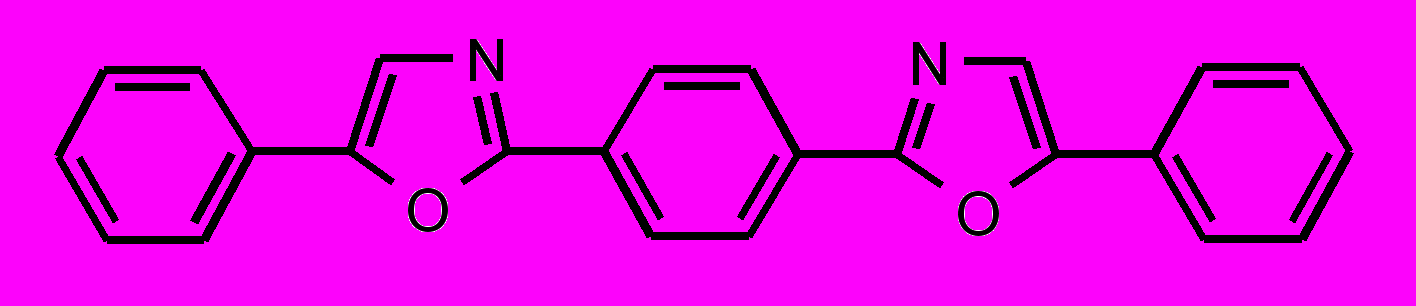 | |
| 39 | 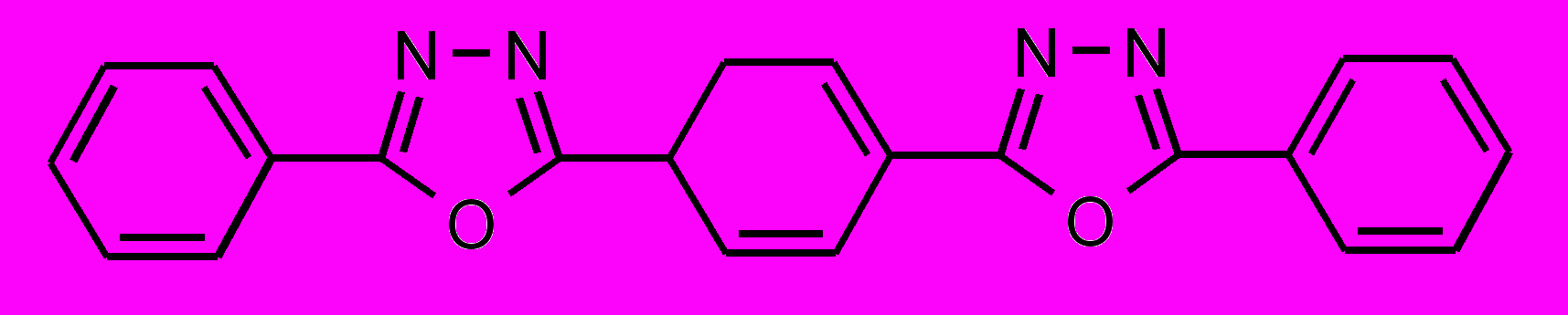 | 40 | 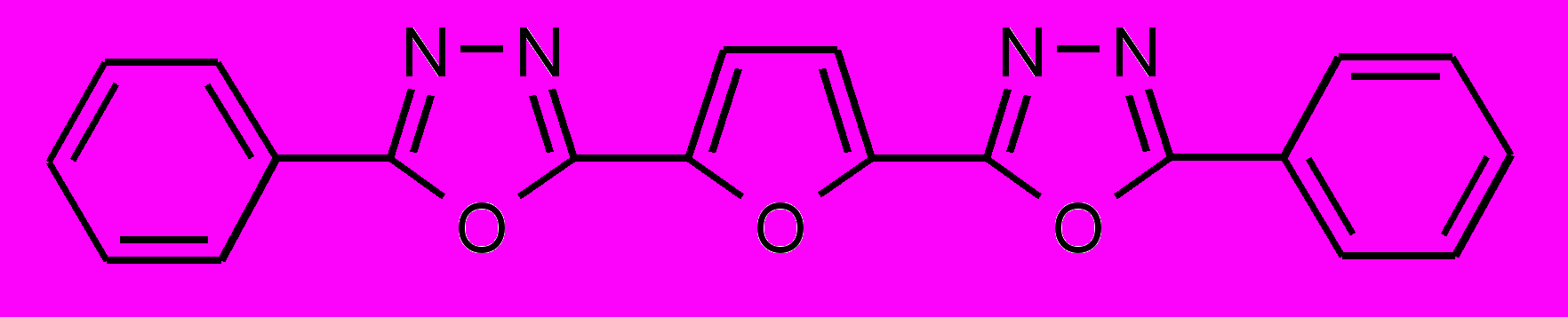 | |
| 41 | 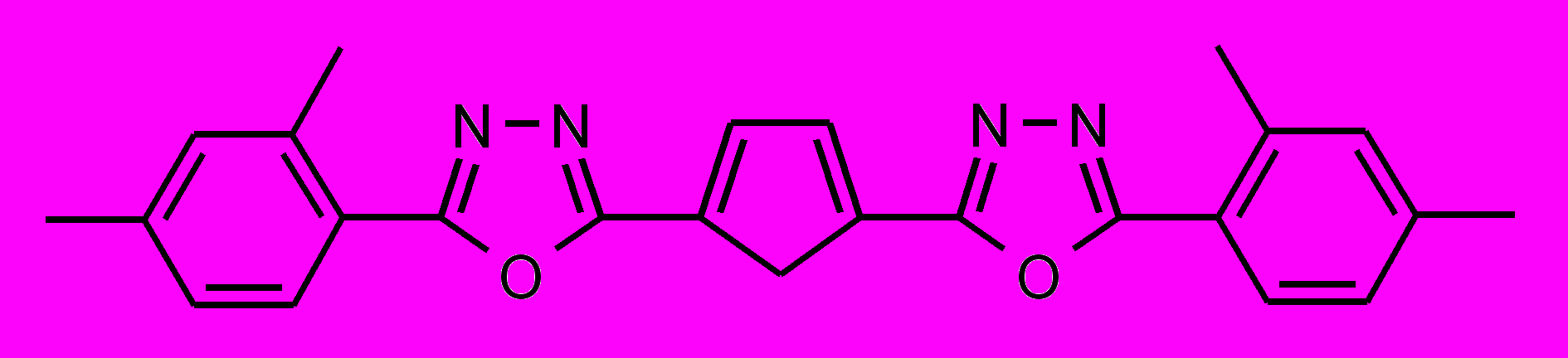 | | |
Наиболее информативным является анализ сопряженных электронно-колебательных спектров сложных молекул с разрешенной колебательной структурой. Для получения тонкоструктурных спектров исследуемых соединений был выбран метод Шпольского.
В процессе работы для исследуемых соединений были измерены спектры флуоресценции, поглощения (при низких температурах – спектры возбуждения флуоресценции) в интервале температур от 293 до 4,2 К, квантовые выходы, кинетика затухания флуоресценции. Для ряда соединений вычислялись константы скоростей излучательного (ки) и безызлучательных (кб) процессов. Исследовались зависимости регистрируемых спектров от концентрации растворов (10-4–10-8 М/л), спектров флуоресценции от длины волны возбуждающего света, спектров возбуждения флуоресценции — от длины волны регистрации.
Практически для всех исследованных соединений методом Шпольского были получены сопряженные спектры флуоресценции и возбуждения флуоресценции при 4.2 К. Для многих соединений (соединения 1, 2, 3, 4, 6, 8, 15, 38, 39, 40, 41) были выполнены исследования колебательных спектров комбинационного рассеяния в порошках и инфра красных спектров, что значительно упростило колебательный анализ тонкоструктурных сопряженных спектров. Спектры соединения 11, 16-21, 29-32 в матрицах Шпольского получить не удалось, но достаточно узкополосные спектры при 77 К были получены в растворах толуола.
Тонкая структура в сопряженных спектрах большинства исследуемых соединений проявлялась в виде пиков на сплошном интенсивном фоне. Положения пиков определяются из экспериментальных спектров однозначно. Определить интенсивность полос непосредственно из сопряженных спектров для большинства исследуемых соединений невозможно. Во всех изученных сопряженных спектрах наблюдается нарушение зеркальной симметрии.
Анализ распределения интенсивности по сопряженным спектрам требует знание вероятности вибронных переходов, т.е. относительных интенсивностей вибронных полос из которых состоят спектры. Для нахождения относительных интенсивностей полос был разработан метод моделирования спектров.
Спектр, как известно, является совокупностью вибронных полос, каждая из которых состоит из бесфононной линии (БФЛ) и фононного крыла (ФК). Для анализа тонкоструктурных спектров нельзя пренебрегать формой полосы, так как относительная интенсивность вибронной полосы пропорциональна площади под огибающей кривой. Зная положение каждой полосы можно моделировать полный вибронный спектр серией вибронных полос. Каждая вибронная полоса представляется суммой узкой бесфононной линии, моделируемой функцией Лоренца, и широкого фононного крыла, моделируемого функцией Гаусса. В этом случае мы упрощаем вид фононного крыла, который в действительности имеет более сложный вид [16]. Но как будет видно в дальнейшем, это не оказывает существенного влияния на окончательный вид спектра. Соотношение интенсивностей БФЛ и ФК в вибронной полосе отражает фактор Дебая-Валлера. Подбирая, в процессе моделирования спектра, параметры, отвечающие за вид вибронной полосы, можно добиться практически полного совпадения полученного таким образом спектра с экспериментальным. Такое моделирование позволяет определить относительные интенсивности измеренных в опыте вибронных пиков, сделать вибрационный анализ сопряженных спектров, и в конечном итоге количественно установить вклад внутримолекулярных взаимодействий в формирование тонкоструктурных сопряженных спектров.
Таким образом, каждая вибронная полоса моделируется суммой узкой БФЛ лоренцевской формы и широкого ФК гауссовой формы (рис. 1). ФК расположено с красной и синей стороны по отношению к БФЛ в спектрах флуоресценции и спектрах возбуждения флуоресценции, соответственно. Ширина БФЛ растет с увеличением номера вибронного уровня, что принимает во внимание уменьшение времени жизни колебательного состояния с ростом его энергии.

Р
 исунок 1. Моделированная электрон-фононная полоса в спектре флуоресценции, состоящая из бесфононной линии (1) и фононного крыла (2).
исунок 1. Моделированная электрон-фононная полоса в спектре флуоресценции, состоящая из бесфононной линии (1) и фононного крыла (2).При моделировании оптимизируются следующие параметры: ширина БФЛ - БФЛ, ширина ФК - ФК, значение фактора Дебая-Валлера -
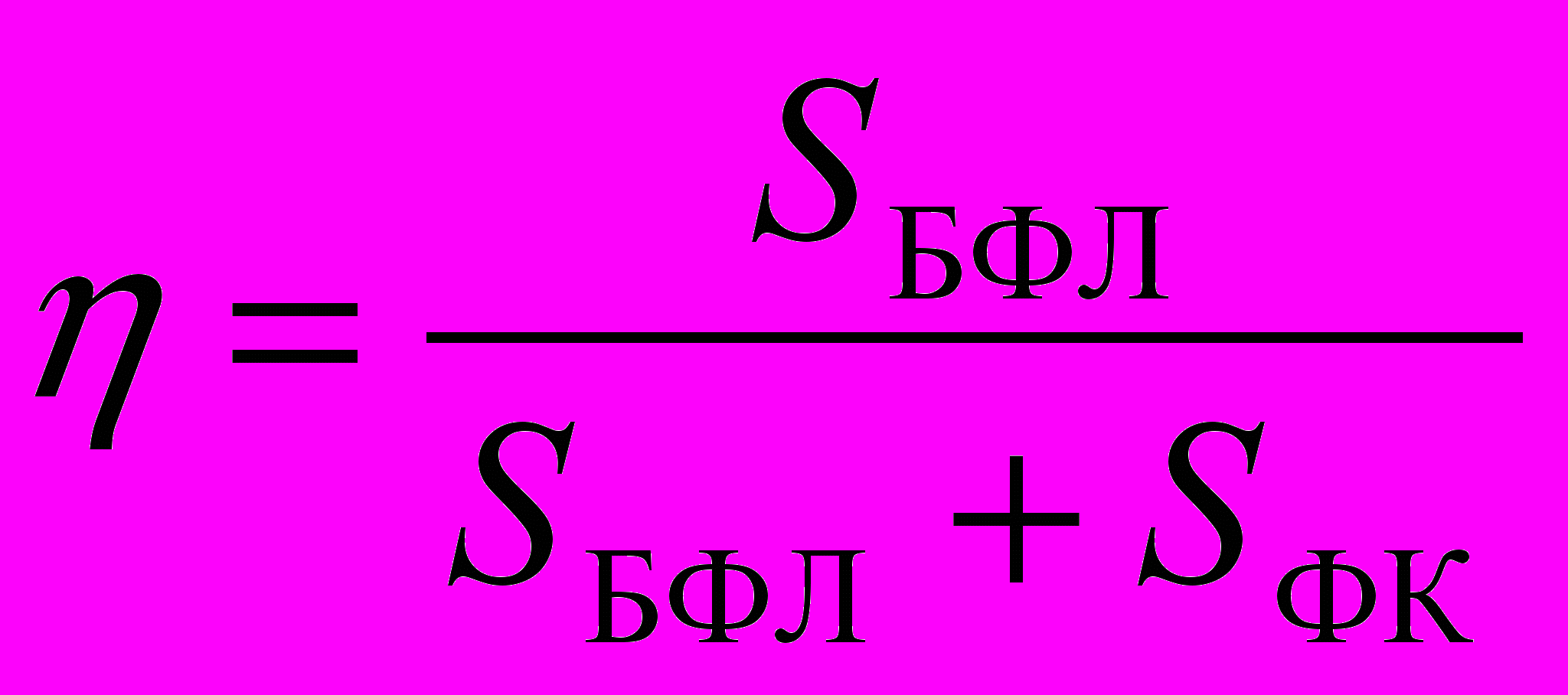 (SБФЛ и SФК – интегральные интенсивности бесфононной линии и фононного крыла, соответственно) и расстояние между максимумами БФЛ и ФК в шкале частот БФЛ ФК так, чтобы при подборе констант вибронного взаимодействия рассчитанные спектры были наиболее близки к экспериментальным. Все параметры определены с учетом неоднородного уширения, поскольку имеющиеся экспериментальные данные не позволяют разделить однородное и неоднородное уширения спектров.
(SБФЛ и SФК – интегральные интенсивности бесфононной линии и фононного крыла, соответственно) и расстояние между максимумами БФЛ и ФК в шкале частот БФЛ ФК так, чтобы при подборе констант вибронного взаимодействия рассчитанные спектры были наиболее близки к экспериментальным. Все параметры определены с учетом неоднородного уширения, поскольку имеющиеся экспериментальные данные не позволяют разделить однородное и неоднородное уширения спектров. Полученные в результате моделирования относительные интенсивности вибронных переходов позволяют рассчитать параметры внутримолекулярных взаимодействий по формулам, полученным для одноквантовых переходов и их обертонов и комбинаций [7,8].
Проверка достоверности рассчитанных параметров проводилась путем расчета интенсивности обертонов и комбинаций частот нормальных колебаний и сравнения полученных величин с экспериментальными (в нашем случае с относительными интенсивностями вибронных полос, полученными в процессе моделирования).
В третьей главе рассмотрены особенности спектрально-люминесцентных свойств органических соединений цепочечного строения. Структурной особенностью молекул соединений, исследуемых в данной работе, является то, что они могут обладать не единственной плоской молекулярной структурой. Поэтому в растворах они могут находиться в виде набора конформеров. Причем неплоские конформеры, как правило, имеют низкий квантовый выход флуоресценции.
Практически все исследуемые вещества плохо растворяются в н-парафинах. Лучше всего эти соединения растворяются в полярных растворителях: толуоле, хлороформе, ДМСО. При замораживании растворов, как правило, при концентрации более 10-4 М/л идет выкристаллизация, и образуются самоассоциаты. В таком случае спектры растворов представляют собой суперпозицию спектров одиночных молекул и спектров различных ассоциатов. В одних случаях они разделены спектрально и спектры ассоциатов не мешают анализу молекулярных спектров (как в случае соединений 14, 15, 24), а в других - спектры ассоциатов и мономолекул перекрываются (соединения 2, 38). Во-вторых, в твердых матрицах могут существовать несколько люминесцирующих конформеров молекул исследуемых соединений.
Изучение кинетики затухания флуоресценции в разных участках спектра показало, что при понижении температуры в растворах многих исследованных соединений, существуют несколько излучающих центров, так как во многих случаях было отмечено, что при 77 К экспоненциальный характер затухания нарушен и может быть аппроксимирован суммой двух и более экспонент.
Ассоциаты, были обнаружены в растворах н-парафинов при 77 и 4,2 К и концентрациях С10-4 М/л у соединений, являющимися представителями всех трех групп исследуемых соединений (табл. 1): у соединения 2 (ДСБ), соединения 14, соединения 15, соединения 24, у гетероциклического соединения соединения 38 (РОРОР). В растворе толуола (слабо полярного растворителя) ассоциаты были обнаружены при низких температурах только у соединения 14 с донорно-акцепторными заместителями.
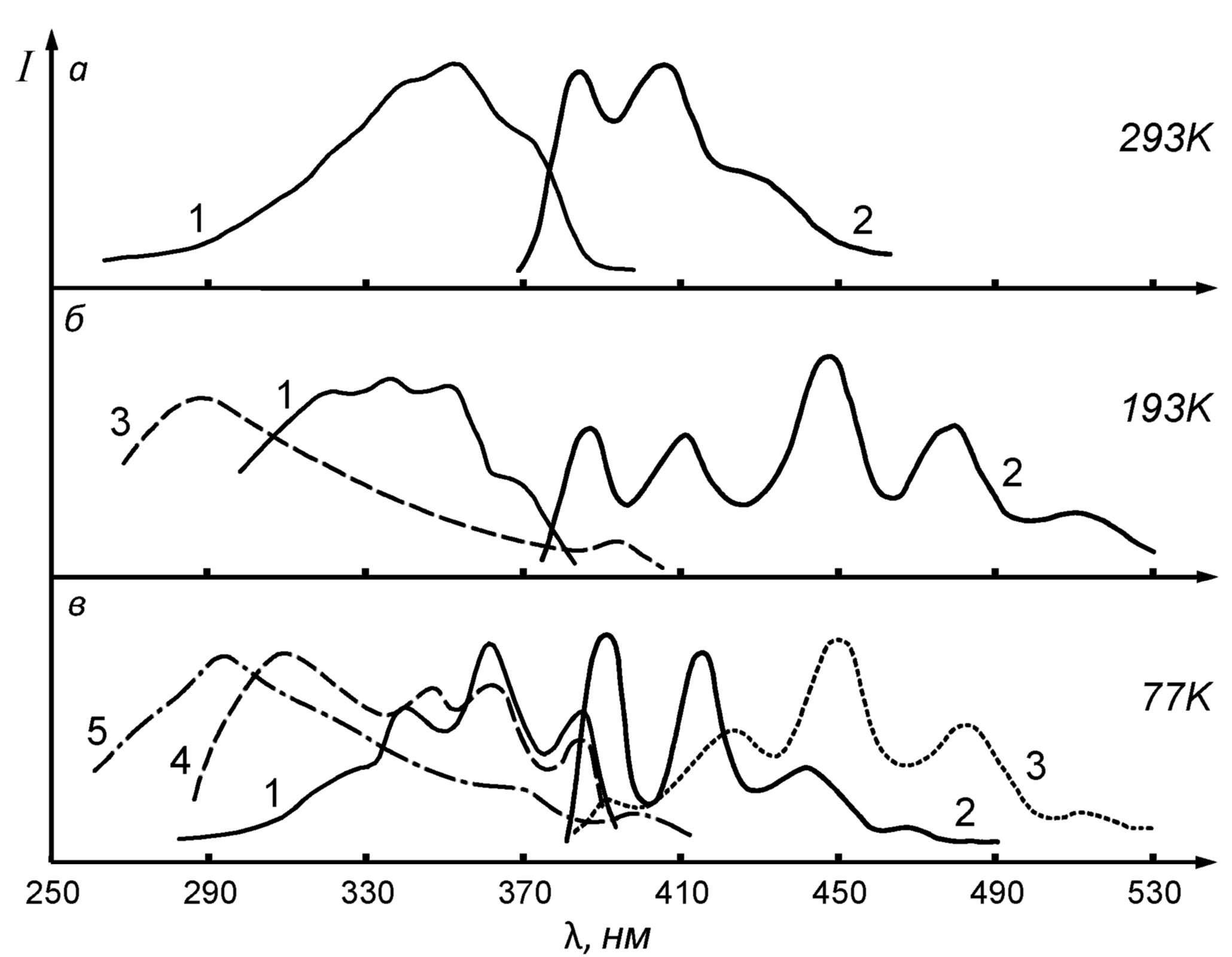
Рисунок 2. Спектры флуоресценции (возб. =360 нм) и возбуждения флуоресценции соединения 2 (ДСБ) в н-гептане, концентрация С=510-5 М/л при разных температурах и скорости замораживания. а) 1 – поглощение, 2 – флуоресценция; б) 1 и 3 – возбуждение флуоресценции рег=410 нм и 480 нм, соответственно, 2 – флуоресценция; в) 1, 4, 5 – возбуждение флуоресценции рег=415 нм, 450 нм, 480 нм, соответственно, 2,3 – флуоресценция; 1, 2, 4 – быстрое замораживание от 293 К, 3,5 – быстрое замораживание от 193 К.
Сопряженные спектры ДСБ в гептане при температуре 193 К (вблизи точки замерзания н-гептана) и 77 К приведены на рис.2. Видно, что понижение температуры приводит к изменению спектров флуоресценции, а именно возрастание интенсивности длинноволновых и уменьшение относительной интенсивности коротковолновых полос.
В растворах с низкой концентрацией (С=210-7 М/л) эти изменения невелики и заметны только при понижении температуры до 77 К. Дальнейшее понижение температуры до 4,2 К приводит к появлению в коротковолновой части спектра квазилинейчатой структуры, а в длинноволновой части наблюдается слабая сплошная полоса. В растворе н-гептана при комнатной температуре затухание флуоресценции экспоненциально с =1,70,1 нс. Существование различны форм молекул ДСБ при низкой температуре и высокой концентрации раствора непосредственно подтверждается кинетическими измерениями. Затухание флуоресценции в коротковолновой части спектра при =390 нм экспоненциально с временем 0=1,2 нс. В более длинноволновой части спектра при =500 нм затухание неэкспоненциально и может быть аппроксимировано суммой экспонент с 1=3,3 нс и 2=15 нс. При промежуточной длине волны =454 нм неэкспоненциальность проявляется еще резче и налагаются три экспоненты с временами 0, 1, 2. Сопоставляя эти результаты со спектральными данными, можно заключить, что время 0 соответствует флуоресценции мономеров, а затухание с временами 1 и 2 – флуоресценции ассоциатов. Наличие двух значений свидетельствует, по крайней мере, о двух различных формах ассоциатов, спектры которых, по-видимому, близки друг к другу.
Принимая во внимание температуру начала образования ассоциатов (200250 К), можно предположить, что энергия связи молекул в ассоциате порядка 150 см-1.
Несмотря на малую энергию связи, смещение электронных спектров ассоциатов относительно спектров молекул довольно значительно. Кроме того, спектры ассоциатов имеют аномально большой стоксов сдвиг. Это можно объяснить наличием у адиабатического потенциала по некоторой нормальной координате двух минимумов. Отметим в заключение, что спектр флуоресцеции ассоциатов, в отличие от спектра мономерных молекул, остается сплошным бесструктурным и при 4,2 К.
П
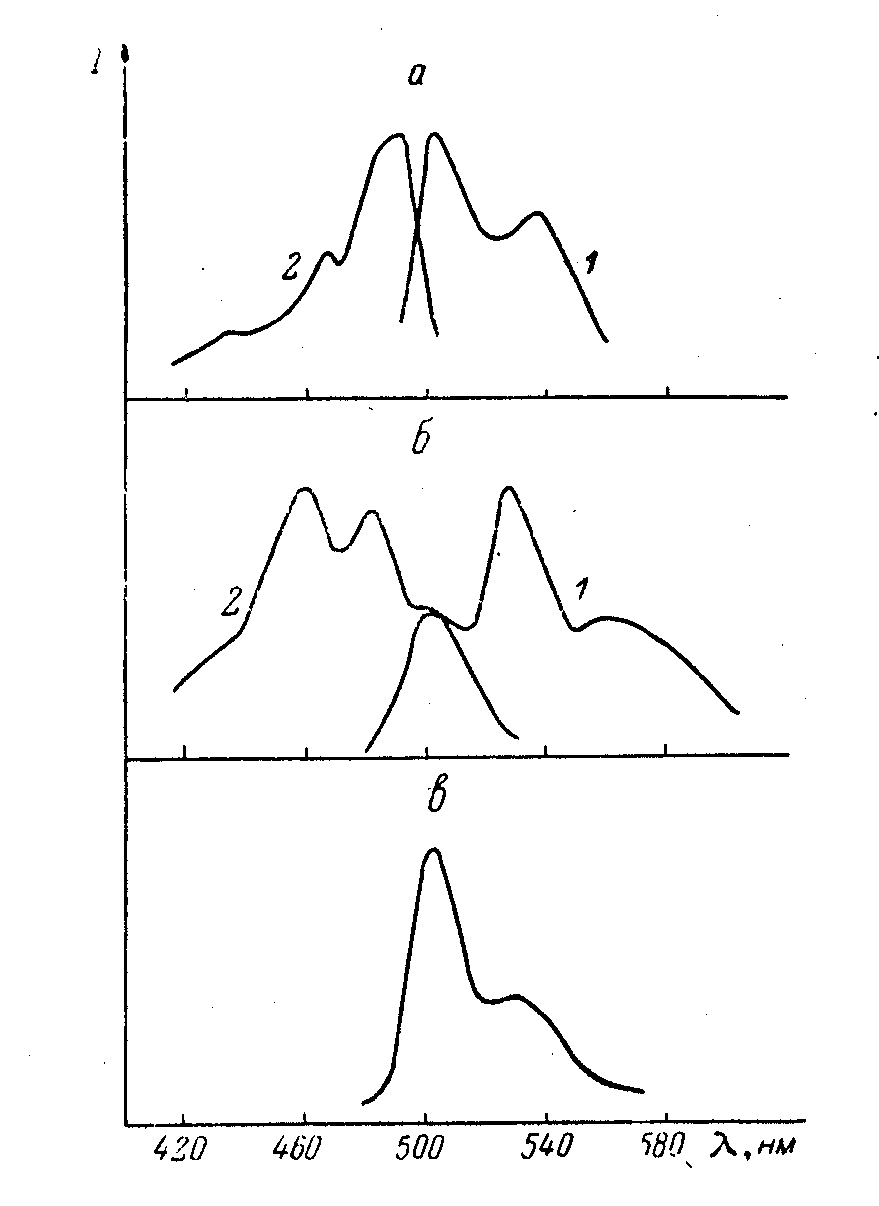 ри комнатной температуре у соединения 14 во всех применявшихся растворителях (н-октане, н-гептане, толуоле, этаноле и диметилформамиде) с концентрацией 10-4 м/л существует один излучающий центр. На рис. 3 приведены спектры флуоресценции и возбуждения флуоресценции в слабо полярном растворителе - толуоле при 77 К. В спектре флуоресценции при возбуждении различными длинами волн наблюдается перераспределение интенсивности между полосами, а при возбуждении 420-470 нм появлялась длинноволновая полоса ~565 нм (рис. 3 б). В спектрах возбуждения флуоресценции при регистрации в различных областях спектра также происходило перераспределение интенсивности между К.
ри комнатной температуре у соединения 14 во всех применявшихся растворителях (н-октане, н-гептане, толуоле, этаноле и диметилформамиде) с концентрацией 10-4 м/л существует один излучающий центр. На рис. 3 приведены спектры флуоресценции и возбуждения флуоресценции в слабо полярном растворителе - толуоле при 77 К. В спектре флуоресценции при возбуждении различными длинами волн наблюдается перераспределение интенсивности между полосами, а при возбуждении 420-470 нм появлялась длинноволновая полоса ~565 нм (рис. 3 б). В спектрах возбуждения флуоресценции при регистрации в различных областях спектра также происходило перераспределение интенсивности между К. Рис. 3. Спектры флуоресценции
и возбуждения соединения 14 в толуоле
при 77 К. Концентрация: а, б – 10-4 М/л, в – 10-6 М/л;
а: 1 – возб = 490 нм, 2 – рег = 510 нм;
б: 1 – возб = 450 нм, 2 – рег = 565 нм;
в: возб = 450, 490 нм.
полосами, а при регистрации в области <~565 нм наблюдалась длинноволновая полоса ~503 - 505 нм. Для растворов же с концентрацией 10-6 М/л, как и для спектров флуоресценции, так и для спектров возбуждения флуоресценции такой зависимости практически не наблюдалось. Спектры и возбуждения флуоресценци также зависят от длины волны регистрации, и от скорости замораживания от комнатной температуры до 77
Спектры флуоресценции соединения 14 в полярных растворителях, таких как этанол и диметилформамид, даже при концентрации 10-4 м/л практически не зависят от длины волны возбуждения, а спектры возбуждения флуоресценции — от области регистрации.
Длинноволновые полосы, присутствующие в спектрах флуоресценции и возбуждения флуоресценции соединения 14 в толуоле с концентрацией 10-4 М/л, вероятней всего принадлежат ассоциатам этого соединения. Коротковолновая полоса в спектре флуоресценции —500 нм относится к мономерной форме этого соединения. Ее вибронным повторением является полоса ~535—537 нм, наблюдавшаяся при возбуждении =490 нм. Интенсивная полоса флуоресценции ~530 нм (рис. 3 б), как и более коротковолновые полосы в спектре возбуждения флуоресценции ~440, 465, 485 нм (рис. 3, а), обусловлены перекрыванием полос мономеров и ассоциатов. В полярных растворителях, таких как этанол и диметилформамид, даже при концентрации 10-4 м/л не наблюдается зависимость спектров от возб и рег.
Аналитически были выделены спектры флуоресценции и возбуждения флуоресценции мономера и спектр флуоресценции ассоциата. Спектр возбуждения флуоресценции ассоциата был измерен при регистрации в области флуоресценции 565—570 нм.
Спектры флуоресценции и возбуждения флуоресценции соединения 14 в толуоле при 77 К практически зеркально симметричны, в то время как между спектрами флуоресценции и возбуждения флуоресценции ассоциатов наблюдается сильное отклонение от зеркальной симметрии. Его нельзя объяснить неадиабатическим взаимодействием, так как следующий электронный переход расположен около 285 нм на расстоянии 13000 см-1 от области исследуемого самого длинноволнового электронного перехода, поэтому влияние второго возбужденного электронного состояния на нижайшее возбужденное электронное состояние должно быть очень мало.
Можно предположить, что подобная незеркальность между сопряженными спектрами ассоциатов соединения 14 может быть по аналогии со спектрами, рассчитанными в работах [14-15] качественно объяснена спектральным проявлением двух минимумов адиабатического потенциала по некоторой нормальной координате в основном состоянии. Излучательный переход попадает в этом случае в область максимума потенциального барьера, конечное состояние является сильно ангармоничным.
В растворах исследуемых кросс-сопряженных кетонов 17, 18, 19, 21, 22 в толуоле случае при 77 К проявляется люминесценция как транс- так и цис- конформера, причем независимо от того как замерзает толуол: образуется стекло или поликристаллическая масса.
Излучение двух конформеров было обнаружено в растворах толуола у восьми соединений с карбонильной группой: 17, 18, 19, 21, 22, 26, 27, 28 (табл. 1). Кинетические измерения показали присутствие двух конформеров в растворе н-октана при 77 К у молекул соединений 26, 27, 28. Наличие карбонильной группы обусловливает существенную полярность кетонов. А, следовательно, и взаимодействие молекул исследуемых кетонов 17, 18, 19, 21, 22 возрастает даже со слабо полярным растворителем, таким как толуол. В этом случае при низких температурах в жестких матрицах проявляется люминесценция как транс- так и цис- конформера, причем независимо от того как замерзает толуол: образуется стекло или поликристаллическая масса.
Два вида поворотных конформеров для компланарных в целом молекул гетероциклических соединений 33-36 цепочечного строения проявляются также и в кинетических измерениях длительности флуоресценции растворов в н-парафинах при 77 К.
В растворах РОРОР (соединение 38) в н-октане анализ концентрационной зависимости спектров флуоресценции позволил сделать вывод о существовании двух мономеров-конформеров - «коротковолнового» тонкоструктурного и «длинноволнового» менее структурного. Из экспериментального спектра флуоресценции были выделены спектры ассоциатов и «длинноволнового» мономера.
Исследование кинетики затухания флуоресценции показали, что для растворов РОРОР в н-октане при комнатной температуре значения t одинаково по всему спектру и равно 1.2 нс. При 77 К наблюдалось свечение двух центров, значения которых равны 1.3 и 7.5 нс. Причем вклад второй компоненты возрастает с продвижением в длинноволновую область. «Длинноволновый» конформер, по-видимому, не проявился из-за очень слабой интенсивности свечения. При изменении концентрации от 10-4 до 10-5 М/л вклад второй компоненты (7.5 нc) с продвижением в длинноволновую область спектра также увеличивается, но в меньшей степени. Следовательно, вторая компонента обусловлена свечением ассоциатов, а первая — структурной флуоресценцией «коротковолнового» конформера. Возможны два конформера РОРОР, отличающихся взаимным расположением оксазольных циклов. Следует отметить, что наличие конформеров у 33-36 и РОРОР обнаруживается только с помощью флуоресцентных измерений.
Спектры флуоресценции и возбуждения флуоресценции РОРОР в н-октане исследовались при концентрации растворов 10-6- 10-4 М/л при 77 и 4.2 К (рис. 3.13, а, б) и была обнаружена зависимость спектров от lвозб и рег.
Уменьшение интенсивности длинноволновой широкой полосы при переходе от концентрации 10-4 к 10-5 и 10-6 М/л при 4,2 К в отличие от интенсивности тонкоструктурных и двух менее структурных полос флуоресценции (рис. 4, б) позволяет сделать вывод, что эта полоса принадлежит ассоциатам, а другие полосы — двум мономерам-конформерам —
«коротковолновому» тонкоструктурному и «длинноволновому» менее структурному. Очень слабая полоса ассоциатов была зарегистрирована в жидком растворе вблизи точки плавления октана. Исходя из того, что при большой концентрации длинноволновая полоса принадлежит только ассоциатам, мы выделили полосу ассоциатов и полосы «длинноволнового» конформера. Выделенные три полосы этого конформера симметричны трем слабо выраженным полосам в спектре возбуждения флуоресценции (lmax=402, 394 и 378 нм). Спектр возбуждения флуоресценции ассоциатов наблюдался в области приблизительно от 410 нм до £330 нм. В длинноволновой области он перекрывается со спектром возбуждения флуоресценции «длинноволнового» конформера, а в коротковолновой — «коротковолнового» конформера.
Спектральные и кинетические исследования показали, что в растворах толуола при концентрации 10-5 М/л при комнатной температуре и для 77 К ассоциаты молекул РОРОР не обнаружены.
Как отмечалось ранее, полоса флуоресценции ассоциатов, расположенная при max=460 нм, появляется при возбуждении в области ~ 400 нм. Следовательно, между спектром флуоресценции и спектром возбуждения флуоресценции (поглощения) ассоциатов в отличие от соответствующих спектров мономеров обоих конформеров существует большой стоксов сдвиг (энергетическая щель) порядка 3500 см-1. Значительный стоксов сдвиг наблюдался нами также для ассоциатов ДСБ (2). В [15] показано, что аномально большой стоксов сдвиг проявляется, когда адиабатический потенциал по некоторой нормальной координате имеет два минимума в возбужденном состояний.
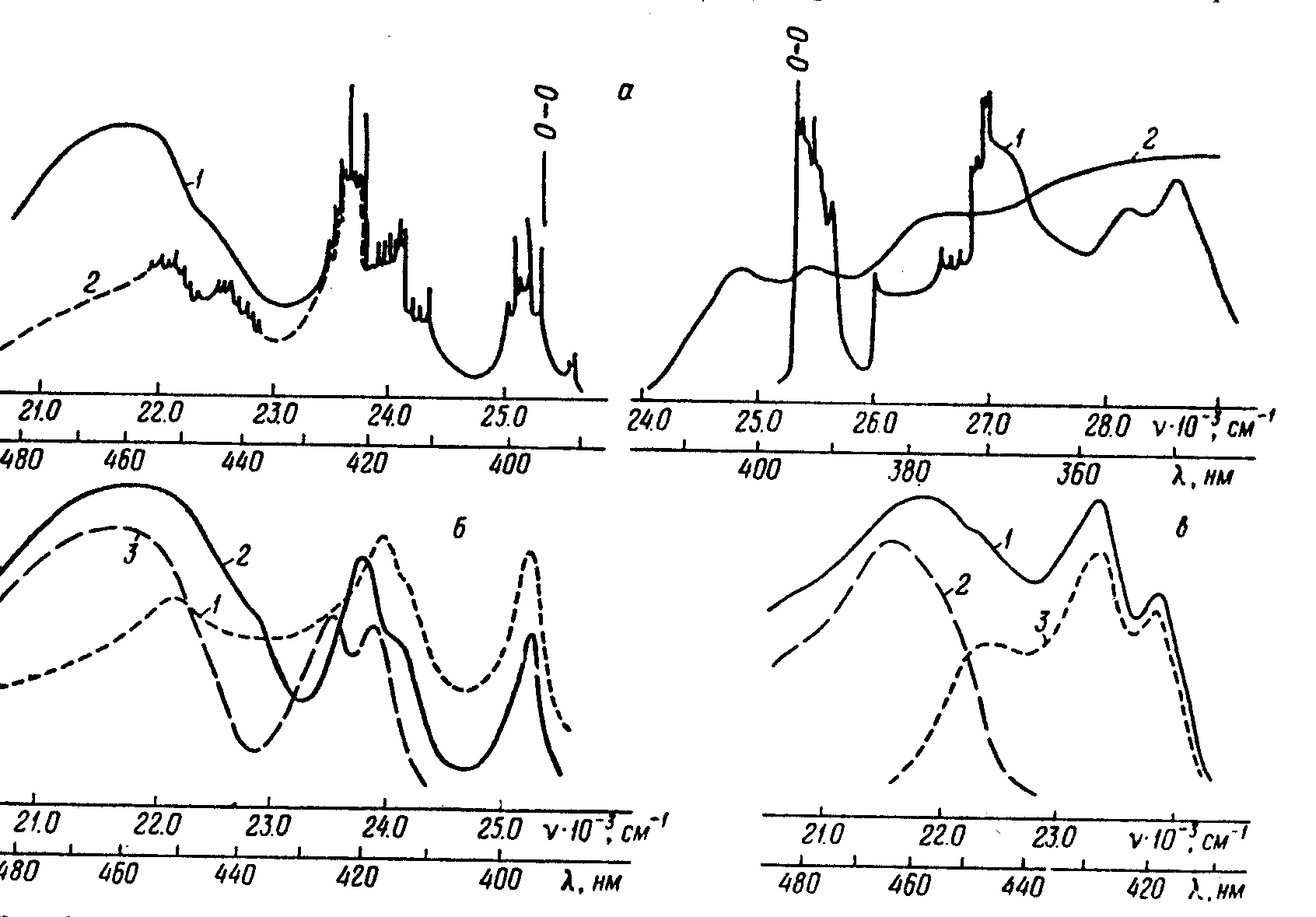
Рисунок 4. Спектры РОРОР в н-октане: а – спектры флуоресценции и возбуждения, концентрация 10-4 М/л, при 4,2 К: возб: 1 – 330 нм, 2 – 370 нм, рег: 1 – 420 нм, 2 – 460 нм; б – спектры флуоресценции при 77 К: возб: 1,2 – 330 нм, 3 – 400 нм, концентрация: 1 – 10-6 М/л, 2,3 – 10-4 М/л; в – спектры флуоресценции при 4,2 К, возб=400 нм, концентрация 10-5 М/л, 1 – суммарный спектр, 2 – спектр ассоциатов, 3 – спектр длинноволнового мономера.
Сделан вывод, что сильная незеркальность между сопряженными спектрами ассоциатов соединений 14 и 38 (РОРОР) может быть проявлением существования двух минимумов адиабатических потенциалов: в основном состоянии у 14 и в возбужденном у 38.
Таким образом, проведенные исследования спектральных свойств ассоциатов и конформеров в растворах соединений, изучаемых в данной работе, позволило найти условия регистрации сопряженных спектров именно мономолекул исследуемых соединений.
В четвертой главе приведен обзор литературных данных по исследованию фото-физических свойств соединений первой группы и результаты собственных исследований их тонкоструктурных спектров. Анализ сопряженных спектров флуоресценции и возбуждения флуоресценции исследованных соединений показал, что замена одного фенильного кольца в молекуле дифенилгексатриена (соединение 5) на мезитильное (соединение 7) не влияет на спектральные свойства, а замена атома водорода на атомы F на двойных связях в молекуле дистирилбензола (соединение 2) приводит к некоторому перераспределению интенсивности по спектрам при практически неизменных колебательных частотах в сопряженных спектрах.
В результате анализа распределения интенсивности по спектру дифенилбутадиена (соединение 4) сделан однозначный вывод о природе нижайшего возбужденного состояния. Наиболее интенсивной полосой в спектре флуоресценции является самая коротковолновая полоса, резонансно совпадающая с самой интенсивной длинноволновой полосой в спектре возбуждения флуоресценции. Следовательно, эта полоса является 0-0 полосой разрешенного по симметрии электронного перехода и нижайшим возбужденным состоянием в этом случае является состояние 11Вu. Запрещенный по симметрии переход 11Ag21Ag лежит в области примерно на 150 - 200 см-1 короче разрешенного перехода.
Вибрационный анализ для молекул дифенилполиенов с числом двойных связей n=3 и 4 возможен только для спектров флуоресценции, так как в спектре возбуждения флуоресценции происходит частичное наложение двух низкоэнергетических электронных переходов. В спектрах исследованных дифенилполиенов, а также в сопряженных спектрах СБ и ДСБ проявляются полосы, обусловленные колебаниями с частотами 1000-1100 см-1 – «дыхательное» колебание бензольного кольца, 1230-1330 см-1 – деформационное колебание боковых связей С-Н полиеновой цепи, 1580-1618 см-1 – колебание С=С двойной связи. Вибронный переход, соответствующий колебанию С=С двойной связи во всех соединений наиболее интенсивный. Полосы, соответствующие этому переходу, сравнимы по интенсивности с полосой 0-0. Валентное колебания полиеновой цепи 1634-1653 см-1 наиболее активно в спектрах стильбена (соединение 1) и дистирилбензола (соединение 2). В сопряженных спектрах соединений этой группы наблюдается незеркальность как в распределении интенсивности, так и по частотам.
Тонкая структура сопряженных спектров флуоресценции и возбуждения флуоресценции ДСБ и ДФБ проявляется на интенсивном сплошном фоне, что делает
а

Рис. 5. Сопряженные спектры флуоресценции (слева) и возбуждения флуоресценции (справа) ДСБ (а- экспериментальные спектры, б – моделированные спектры)
б
невозможным непосредственное измерение относительной интегральной интенсивности вибронной полосы, а, следовательно, количественный анализ внутримолекулярных взаимодействий. Поэтому было проведено моделирование этих спектров по методике, описанной в главе 2. Путем подбора необходимых параметров удалось добиться того, что экспериментальные и моделированные спектры практически совпали. Такое совпадение получено