
Тонкоструктурные спектры и электронно-колебательные взаимодействия сопряженных молекул цепочечного строения
| Вид материала | Автореферат |
СодержаниеТаблица 2. Параметры FC- и HT- взаимодействий в молекулах ДСБ и FДСБ. В пятой главе Шестая глава |
- Реферат Отчет, 51.81kb.
- Исследование процессов взаимодействия молекул водорастворимого фуллерена с магнитными, 37.88kb.
- Программа дисциплины дпп. Ф. 02 Строение молекул и основы квантовой химии, 160.77kb.
- Молекулярная структура вещества. Скорости газовых молекул, 140.08kb.
- Физические величины, измеряемые в аэрогидромеханике и теплофизическом эксперименте., 39.57kb.
- 1. Некоторые вопросы строения веществ, 1501.26kb.
- Программа Государственного экзамена по подготовке магистра по направлению «Радиофизика», 49.35kb.
- Курс лекций «Основы радиоэлектроники» Часть Сигнал и его свойства. Линии передачи, 33.21kb.
- I. Основы физических процессов в ядерных реакторах, 559.27kb.
- Стереохимия, область химии, изучающая пространственное строение молекул и влияние этого, 19.63kb.
Таблица 2. Параметры FC- и HT- взаимодействий в молекулах ДСБ и FДСБ.
Примечание. В скобках приведены частоты колебаний возбужденного состояния при следующих параметрах: ширины БФЛ для обоих соединений равны 20 см-1, ширины ФК – 120 см-1, величина фактора Дебая-Валлера равна 0.15. Таким образом, стало ясно, что фон, на котором проявляются вибронные пики обусловлен именно интенсивными ФК, а не неоднородным уширением. Был проведен расчет параметров внутримолекулярного взаимодействия для 11 нормальных колебаний молекул СБ и 5-ти нормальных колебаний молекулы ДФБ, 33-ти нормальных колебаний молекулы ДСБ и 25-ти — для молекулы FДСБ (таблица 2). Число нормальных колебаний у молекул этой группы соединений существенно больше, чем у «жестких» молекул, в спектрах которых проявляется не более 10-ти нормальных колебаний [17-19], причем в большинстве случаев расчет параметров показывает, что та или иная полоса в вибронном спектре «жесткой» молекулы проявляется только за счет FC- или НТ- взаимодействий. То, что для молекулы ДФБ параметры были рассчитаны только для пяти нормальных колебаний объясняется тем, что для анализа используется только спектр флуоресценции. Достоверность полученных величин FC- и НТ- параметров подтверждается расчетом относительных интенсивностей полос, соответствующих обертонам и комбинациям нормальных колебаний. В сопряженных спектрах исследуемых молекул практически для всех нормальных колебаний превалирует FC-взаимодействие, но влиянием НТ-взаимодействия на формирование спектров пренебрегать, как это делают авторы [20], для молекул дифенилполиенов, нельзя. Незеркальность между сопряженными спектрами этой группы соединений обусловлена интерференцией FC- и НТ-взаимодействий. В пятой главе приведены обзор литературных данных по фото-физическим свойствам и результаты исследования сопряженных спектров соединений второй группы, молекулы которых являются замещенными или производными от молекул соединений первой группы. Все соединения этой группы впервые синтезированы. Сопряженные спектры флуоресценции и возбуждения флуоресценции замещенных полиенов и дифенилполиенов с числом двойных связей в цепи n = 2 или n = 3 (заместители — NO2, N(CH3)2, NH2, CN) были измерены в «жестких» парафиновых матрицах при 4.2 К. Тонкая колебательная структура в спектрах возбуждения флуоресценции как и в спектрах флуоресценции всех замещенных дифенилбутадиенов, в отличие от дифенилбутадиена проявляется от полосы чисто электронного перехода до 3500 - 4000 см-1. В этих условиях в сопряженных спектрах соединений 10, 13 и 14 содержащих разнотипные заместители, наблюдается значительный сдвиг в низкочастотную область по сравнению с аналогичными спектрами дифенилбутадиена (~8000 см 1). Такой сдвиг спектров может быть объяснен эффектом сопряжения заместителей с различными электронно-донорными N(CH3)2 и электронно-акцепторными NH2 свойствами. А в сопряженных спектрах соединений 8, 9, имеющих только один электронно-донорный заместитель, этот сдвиг гораздо меньше (~2200 см-1 для 8, ~3500 см-1 для 9). Спектры флуоресценции и возбуждения флуоресценции соединения 12, содержащего в параположении молекулы электронно-акцепторный заместитель NO2, испытывают аналогичный сдвиг ~2500 см-1 по сравнению со спектрами 1,6-дифенилгексатриена-1,3,5. Следовательно, влияние заместителей на спектральные свойства соединений 8, 9, 12 меньше, чем у соединений 10, 13 и 14. В спектрах замещенных гексатриена с разнотипными заместителями, но без донорно-акцепторного взаимодействия наблюдалось значительно меньшее длинноволновое смещение по сравнению с данными спектров ДФГ, чем у 10, 13 и 14. Наибольший квантовый выход флуоресценции в молекуле 10 по сравнению с квантовыми выходами соединений 8,9,12 может быть также обусловлен сильным электронным донорно–акцепторным взаимодействием. Спектры соединений 9 и 8 отличаются от спектров 10 и 12 значительно более ярко выраженной колебательной структурой. Полоса чисто электронного 0-0 перехода молекулы 8 по сравнению с молекулой 9 смещена в коротковолновую область примерно на 1300 см-1, что, по всей видимости, связано с заменой заместителя N(CH3)2 в молекуле 9 на NH2 в молекуле 8. Сильное донорно-акцепторное взаимодействие в молекуле диена (соединение 13) ослабляется в триене (соединение 14), и спектр триена приобретает черты спектра полиенового соединения. Это подтверждается также данными КР. Можно сделать вывод, что наиболее длинноволновый электронный переход в молекуле триена в отличие от диена локализован главным образом на полиеновой цепи. Эти выводы соответствуют также данным спектра РКР триена. При возбуждении в области длинноволнового электронного перехода молекулы триена (450515 нм) резко возрастает интенсивность линии полносимметричного колебания С=С связи 1540 см-1. Исследование спектральных свойств впервые синтезированных кросс-сопряженных кетонов значительно усложняет плохая растворимость этих соединений в н-парафинах. Далеко не для всех соединений с кето-группой в структуре молекулы удалось зарегистрировать тонкоструктурные спектры. Спектральные исследования соединений 15-17 (полиеновых бис-,-диметиламинокетонов) показывают, что даже небольшие изменения в структуре приводят к значительным отличиям в их спектральных свойствах. Молекула соединения 15 имеющая в своей структуре четыре двойных связи симметрична относительно центральной кетогруппы, в молекуле соединения 16 при том же числе двойных связей эта симметрия нарушена. Структура 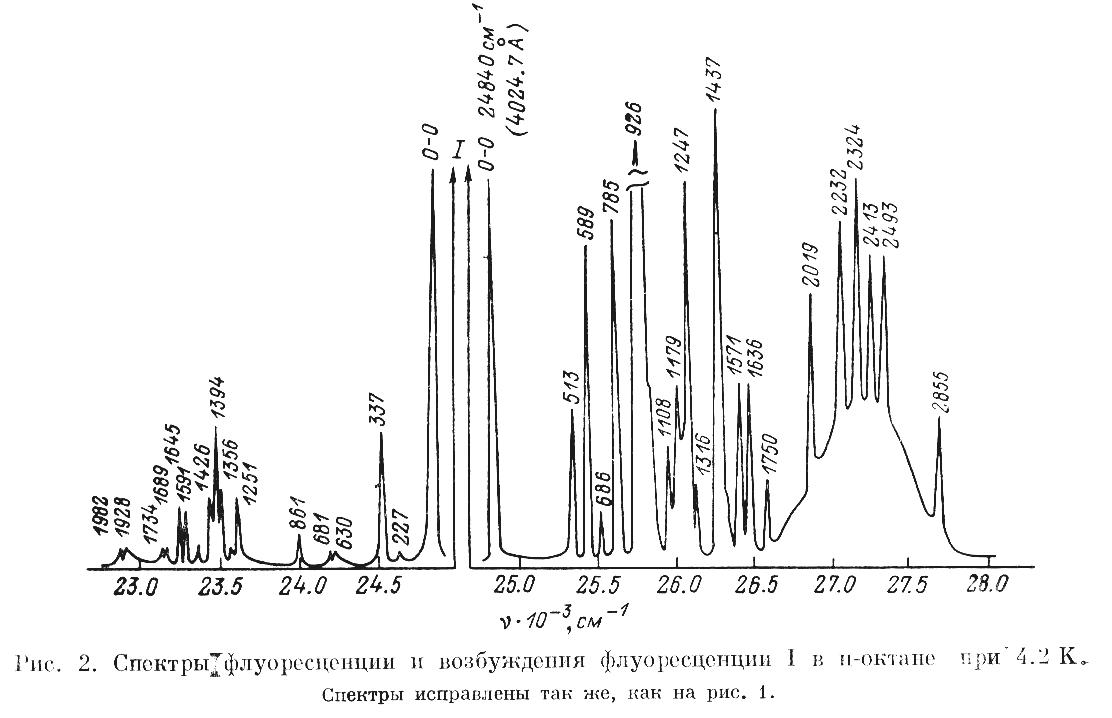 Рисунок 6. Спектры флуоресценции (слева) и возбуждения флуоресценции (справа) соединения 15 в н-октане при 4,2 К. молекулы соединения 17 отличается от 15 наличием центрального шестичленного цикла. Это приводит к тому , что сопряженные спектры соединения 15 в октане при 4,2 К квазилинейчатые, а спектры соединения 16 и 17 – широкополосные, что еще раз подтверждает необходимость «удобного» встраивания молекулы примеси в матрицу растворителя. Между спектрами флуоресценции и возбуждения флуоресценции соединения 15 в н-октане при 4,2 К наблюдается очень сильное отклонение от зеркальной симметрии как по распределению интенсивности, так и по колебательным частотам. В спектре возбуждения флуоресценции происходит наложение двух спектров: нормального, полосы которого имеют аналоги в спектре флуоресценции, и «аномального», не имеющего соответствующего спектра флуоресценции. Для объяснения этой аномалии в спектре возбуждения флуоресценции мы применили модель двухъямного адиабатического потенциала по некоторой внутримолекулярной координате в возбужденном состоянии. В этом случае спектр возбуждения флуоресценции можно разделить на две компоненты. Одна из них «нормальная». К ней относятся полосы, у которых наблюдаются обертоны и, главное, имеются аналоги в спектре флуоресценции. Она обусловлена обычным одноямным потенциалом. Вторая – «аномальная» компонента спектра связана с проявлением двухъямного адиабатического потенциала. Полоса 926 см-1 расположенная в этой области, вероятно, соответствует ангармонической колебательной моде двухъямного потенциала. В «аномальной» части спектра расположены полосы, не имеющие аналогов в спектре флуоресценции, например с частотой 589 см-1. Причем её отсутствие нельзя объяснить интерференцией франк-кондоновского и герцберг-теллеровского взаимодействий, как это можно сделать для полос 513 и 785 см-1 в спектре флуоресценции. Спектр возбуждения флуоресценции сильно ангармоничен. Согласно теории предложенной И.С. Осадько [14,15], адиабатический потенциал моделируется выражением U(x) = -2 (cosX - cos 2X - sin X), где X – координата, вдоль которой потенциал имеет два минимума, параметр – передает общую глубину потенциала, – высоту барьера, – ассиметрию потенциала. Были подобраны значения параметров и . При =600 и ==0 получается простой одноямный адиабатический потенциал основного состояния. При значениях параметров =600, =0,37 и =0,1 вычисленный спектр примерно совпадает с экспериментальным. За базисную (нормировочную) линию взята частота 926 см-1. В соединениях 23-25 образуется внутримолекулярная водородная связь. Сопряженные спектры со слабо выраженной тонкой структурой получены для соединений 23 и 25. Введение в структуру молекулы соединения 23 вместо одной двойной связи с электронно-донорным заместителем N(CH3)2 метилпиррольного цикла приводит к большему разрешению колебательной структуры, но и к увеличению интенсивности сплошного фона. Измерение относительной интенсивности вибронных полос по этим спектрам невозможно. У соединения 24 в растворе н-октана, молекула которого является структурной частью как 23, так и 25 сопряженные спектры широкополосные, практически бесструктурные. Сильное нарушение зеркальной симметрии наблюдается в сопряженных спектрах растворов соединения 26 в октане при низких температурах. В то время как в спектре флуоресценции проявляется тонкая структура, спектр возбуждения флуоресценции состоит из широких, бесструктурных полос. Эта особенность сопряженных спектров с вполне приемлемой точностью укладывается в рамки теории двухямных потенциалов в основном и возбужденном состояниях Адиабатические потенциалы основного ( 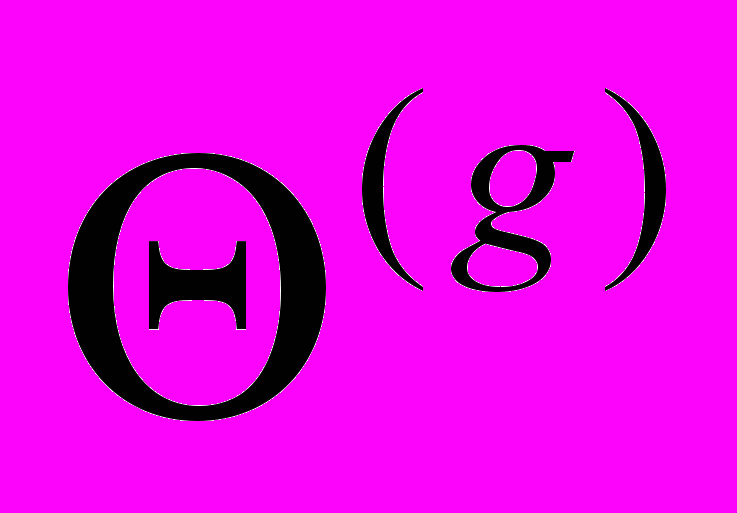 = 3800; = 3800;  = 0.40; = 0.40; 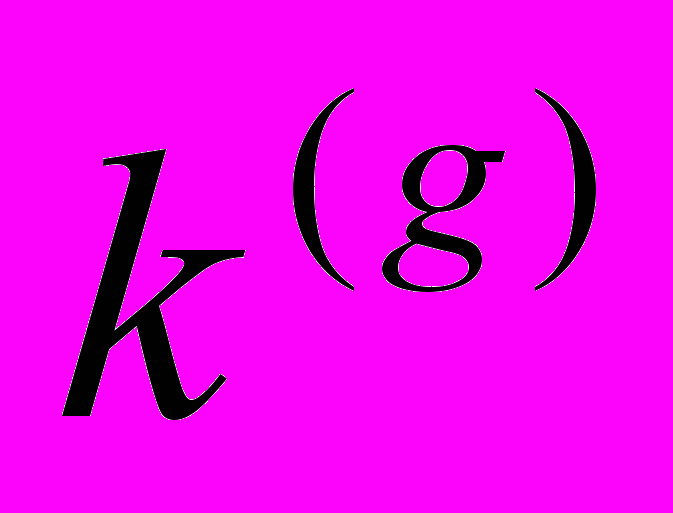 = -0.05) и возбужденного ( = -0.05) и возбужденного (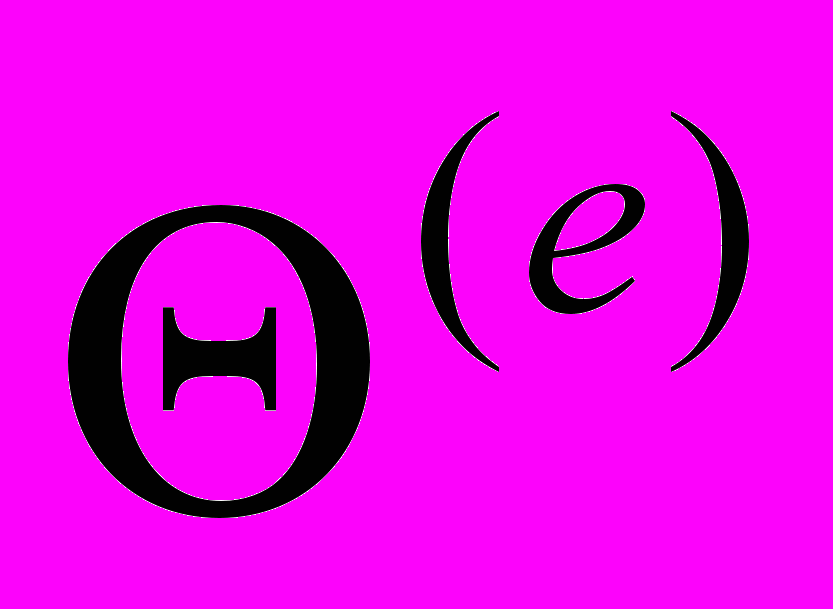 = 1900; = 1900; 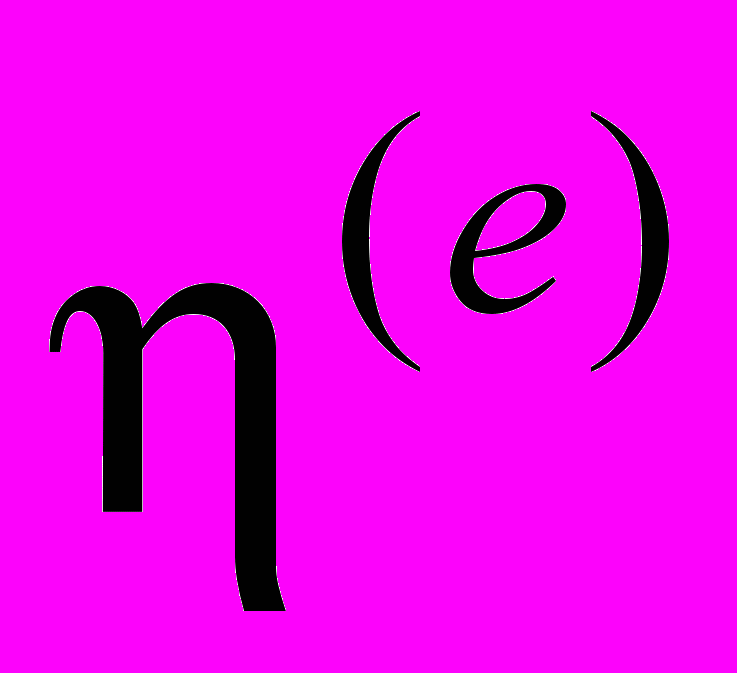 = 0.75; = 0.75; 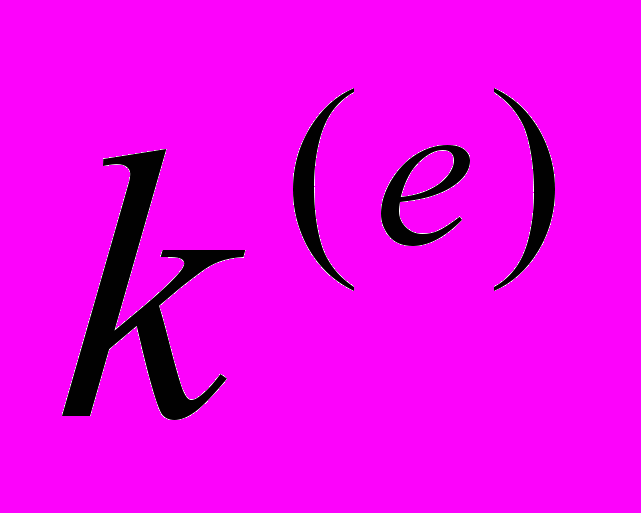 = 0.10) состояний, соответствующие наилучшему согласию с экспериментальными спектрами [21]. = 0.10) состояний, соответствующие наилучшему согласию с экспериментальными спектрами [21].Изменение в структуре молекул 29-32 практически не влияют на спектральные свойства. Сопряженные спектры этих соединений при 77 К широкополосные и практически зеркально симметричные. Спектры соединений 29-31 состоят их трех полос, шириной 350-400 см-1 и лежат в одной области. Наименее структурны спектры соединения 32 в которых проявляются только две полосы. Замена одного из метилпиррольных циклов в молекулах 29 и 30 приводит к длинноволновому смещению сопряженных спектров на 500 см-1. Шестая глава посвящена исследованию тонкоструктурных спектров соединений цепочечного строения, молекулы которых состоят из пятичленных гетероциклов (третья группа соединений). Исследование спектрально-люминесцентных свойств таких соединений при низких температурах в жестких матрицах ранее не проводилось. Результаты исследований спектров пятизвенных цепочечных молекул свидетельствуют о том, что степень разрешенности вибронной структуры спектров флуоресценции и возбуждения флуоресценции чувствительны к концентрации их растворов и температуры охлаждения. Наилучшее разрешение удается достигнуть при 4,2 К для наиболее разбавленных растворов (C 10-6 М/л). Измерены спектры флуоресценции и возбуждения флуоресценции впервые синтезированных соединений, состояших из трех гетероциклов 33-36 в н-гексане а также соединения 37 (РОР) при 77 и 4,2 К. У всех соединений 33-37 тонкая структура в сопряженных спектрах проявляется на интенсивном фоне. Наличие в растворе нескольких конформеров исследуемых соединений, спектры которых налагаются друг на друга, делает невозможным анализ полученных спектров. Для соединений 33-36 наблюдалась зависимость спектров флуоресценции от возб, а спектров возбуждения флуоресценции - от рег. Такие зависимости для сопряженных спектров РОР не обнаружены. Это согласуется с тем, что молекула РОР не может иметь конформеров при компланарности молекулы в целом. Спектр флуоресценции РОР как положению полос, так и по распределению интенсивности в них близок к соответствующему спектру соединения соединения 33 (РОО). Практически совпадают у них положения чисто электронного перехода (полосы 0-0). Для РОР 0-0=29464 см-1, а для РОО 0-0=29427 см-1. Следовательно, замена фенильного кольца на оксазольный цикл в молекуле соединения 33 мало изменяет положение низкоэнергетического возбужденного электронного состояния. Самой интенсивной полосой в сопряженных спектрах является резонансная полоса, соответствующая чисто электронному переходу (полоса 0-0). Сопоставление спектров соедиений 38 (РОРОР) и 39 (PDPDP) показывает, что у соединения с оксадиазольными звеньями спектры смещены на 40 нм в более коротковолновую сторону по сравнению со спектрами соединения с оксазольными звеньями. В отличие от люминофоров РОРОР и PDРDP, спектральные свойства недавно впервые синтезированных соединений 40 и 41 (PDFDP и XDFDX, соответственно) до настоящей работы практически не были изучены. Замена центрального Р-звена на цикл F в молекуле PDРDP приводит к батохромному смещению полосы поглощения на 8 нм и еще большему смещению полосы флуоресценции на 16 нм. Введение по две метильные группы в каждое из концевых Р-колец вызывает дополнительное батохромное смещение. Колебания со значениями частот диапазонов 990-1010 и 1600-1622 см-1 проявляются в спектрах многих производных бензола. Их принято относить к валентным колебаниям бензольного кольца. Колебания с частотами 1176-1196 и 1442-1459 см-1 принято считать характеристичными для сжатия-растяжения одинарных С–С связей между звеньями молекулы. Колебания с частотами 952-961см-1 и 1514-1523 см-1 являются, по-видимому, характеристичными для пятичленных гетероциклов. В молекуле PDFDP эти частоты не проявляются, в то время как в молекуле XDFDX есть достаточно интенсивные вибронные полосы: в спектре возбуждения флуоресценции, соответствующая нормальному колебанию с частотой 930 см-1, и в спектре флуоресценции — с частотой 1523 см-1. В вибронных спектрах всех четырех молекул – PDFDP, XDFDX, РОРОР и PDPDP – присутствуют полосы с частотами 1621-1633 см-1. Замена центрального бензольного цикла в молекуле PDPDP на фурановый в молекулах PDFDP и XDFDX приводит к некоторому изменению частот, как в спектрах КР, так и сопряженных спектрах люминесценции. Так, при этом колебания с частотами 983, 1013, 1113 и 1276 см-1, проявляющиеся в спектрах КР PDFDP, не удается зарегистрировать в спектрах флуоресценции этого соединения, а в сопряженных спектрах XDFDX они проявляются. Как было показано, для всех соединений, состоящих из пяти гетероциклов тонкая структура в сопряженных спектрах проявляется на интенсивном фоне. Поэтому невозможно по экспериментальным спектрам измерить интенсивности вибронных полос. Было проведено моделирование спектров с помощью методики, описанной в главе 2. Моделированные спектры практически совпали с экспериментальными. Такое совпадение получено при следующих параметрах: ширины БФЛ (ГБФЛ): POPOP – 20 см-1, PDPDP – 25 см-1, PDFDP – 40 см-1, XDFDX – 35 см-1; ширины ФК (ГФК): 300 см-1 для POPOP и PDPDP, 414 см-1 для PDFDP и 420 см-1 для XDFDX; фактора Дебая-Валлера (): 0.16 для PDFDP, 0.2 для POPOP, PDPDP и XDFDX. Значение ГБФЛ для всех исследованных молекул этих соединений не превышает 40 см-1, фактор Дебая-Валлера - 0,2. Видно, что ширина БФЛ при моделировании, а, следовательно, неоднородное уширение, у соединений в структуре которых имеются только фенильные циклы в 1,5-2,0 раза меньше, чем у соединений, в молекулах которых центральный цикл – фурановый. Оптимизированные значения ГБФЛ и ГФК свидетельствуют, что довольно интенсивный фон, на котором проявляются вибронные пики, обусловлен именно наложением ФК, а не существующим неоднородным уширением. Большая ширина и значительная интенсивность фононных крыльев свидетельствует о сильном электрон-фононном взаимодействии примесных молекул исследованных соединений с окружающей средой. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||