
Курсовая работа Выделение ароматических углеводородов методами изомеризации и деалкилирования
| Вид материала | Курсовая |
- Твердофазная люминесценция полициклических ароматических углеводородов в условиях адсорбционного, 262.95kb.
- Бактерии-деструкторы ароматических углеводородов и их хлорпроизводных: разнообразие,, 940.42kb.
- М. В. Ломоносова Химический факультет Кафедра аналитической химии сорбция ароматических, 219.97kb.
- Задание егэ по теме «Углеводороды» а задания, 45.66kb.
- «Взаимосвязь предельных, непредельных и ароматических углеводородов», 11.54kb.
- Вопросы для подготовки к зачету по дисциплине: «Эксплуатационные материалы», 51.34kb.
- Свистунова наталья Юрьевна биологические особенности лекарственных и ароматических, 416.08kb.
- Модуль Предельные углеводороды, 224.21kb.
- Ароматические углеводороды. Бензол представитель аренов. Строение молекулы и физические, 54.51kb.
- Университет Кафедра «Химия, химическая технология и экология», 98.9kb.
Курсовая работа
Выделение ароматических углеводородов методами изомеризации и деалкилирования
Содержание
стр.
Введение 3
1. Теоретические основы процесса 4
2. Способы получения ароматических углеводородов 7
3. Сравнение методов получения ароматических углеводородов 9
4. Технология процесса получения ароматических углеводородов 12
5. Оценка методов получения ароматических углеводородов 14
Список литературы 15
Введение
Первые органические вещества, с которыми познакомился человек, были выделены из растительных и животных организмов или из продуктов их жизнедеятельности. Каждый растительный или животный организм представляет собой своеобразную химическую лабораторию в которой протекает множество сложнейших реакций, приводящих к образованию огромного числа органических веществ, как весьма простых (например, метан, муравьиная, щавелевая кислоты и т. п.), так и самых сложных (например, алкалоиды, стероиды, белки).
Характерной чертой органического синтеза у растений является накопление потенциальной химической энергии путем превращения в нее энергии солнечных лучей. С помощью хлорофилла на свету растения синтезируют сложнейшие органические соединения из самых простых химических веществ, в конечном счете, из двуокиси углерода, улавливаемой из воздуха, из воды и из минеральных солей, находящихся в почве. По всей вероятности, первичными продуктами фотосинтеза являются углеводы, которые в дальнейшем превращаются в жиры и белковые вещества растительных организмов. Фотосинтез у растений сопровождается выделением кислорода, который, как теперь точно установлено, образуется не из двуокиси углерода, а из воды. Таким образом, путем фотосинтеза в растениях происходит накопление сложных органических веществ. Естественно, что растения в большей мере, чем животные, служат первоисточником получения органических веществ.
Особенно богатым источником органических веществ являются древесные растения.
Наиболее давно известны термические методы переработки древесины, приводящие к разрушению содержащихся в ней сложных органических веществ с образованием более простых соединений.
1. Теоретические основы процесса
Аренами, или ароматическими углеводородами, называют вещества, в молекулах которых содержится одно или несколько бензольных колец.
Простейшим представителем класса Аренов является бензол C6H6.
1. Гомологический ряд, изомерия и номенклатура
Гомологи бензола содержат бензольное кольцо с одним или несколькими алкильными заместителями. Общая формула Аренов C6H2n-6.
СН3
Структурная изомерия гомологов бензола связана со строением и взаимным расположением углеводородных заместителей в бензольном кольце. Изомерия возможна с третьего члена гомологического ряда. Углеводороду состава C8H10 могут отвечать четыре ароматических углеводорода:
СН2-СН3





СН3
1
2
3
4
Этилбензол
1,2 – диметилбензол (о – ксилол)
СН3
СН3
1
1
2
2
СН3
3
3
4
4
1,3 - диметилбензол
(м – ксилол)
СН3
1,4 - диметилбензол
(п – ксилол)
В основу названия аренов положено слово бензол, все гомологи рассматриваются как его производные. Название второго представителя гомологического ряда аренов – метилбензол. Широкое распространение имеют и тривиальные названия. Так, метилбензол называют толуолом. Если заместителей 2 и более, то их перечисляют в порядке старшинства с указанием положения в ядре. Для этого атомы углерода цикла нумеруют, начиная с того, который содержат самый простой заместитель, в том направлении, при котором сумма цифр в названии наименьшая.
Для обозначения взаимного положения двух заместителей в ядре введены особые термины, заимствованные из греческого языка. Для расположенных рядом заместителей используют приставку орто– (orthos – прямой), для находящихся через один атом углерода – мета- (meta – после), для находящихся напротив – пара- (para – напротив). Это полезные термины, их желательно запомнить и не путать. Таким образом, существуют три изомерных ксилола (диметилбензола) – о – ксилол, м – ксилол, п – ксилол.
Общеприняты тривиальные названия самых распространенных радикалов ароматических углеводородов: фенил C6H5– и бензил C6H5-CH2-.
2. Физические свойства
Первые члены гомологического ряда бензола – бесцветные летучие жидкости с характерным запахом, плотность меньше 1 г/мл, в воде они практически не растворяются, однако сами являются хорошими растворителями органических веществ. В гомологическом ряду с увеличением молекулярной массы углеводорода увеличиваются его температуры кипения, плавления и плотность. Арены весьма токсичны, и работа с ними требует строгого соблюдения мер безопасности.
3. Химические свойства
Бензол и его гомологи существенно отличаются по химическим свойствам, как от предельных, так и от непредельных углеводородов. Характерной чертой непредельных углеводородов является их склонность к реакциям присоединения с образованием насыщенных соединений, а также к окислению. Циклическое сопряжение
 -электронов в ароматических углеводородах энергетически выгодно, в результате этого возрастает устойчивость молекулы. Арены с трудом окисляются и восстанавливаются. Будучи формально ненасыщенными соединениями, они не склонны к присоединению, характерными для них являются реакции замещения, при которых сохраняется ароматическая система.
-электронов в ароматических углеводородах энергетически выгодно, в результате этого возрастает устойчивость молекулы. Арены с трудом окисляются и восстанавливаются. Будучи формально ненасыщенными соединениями, они не склонны к присоединению, характерными для них являются реакции замещения, при которых сохраняется ароматическая система. 1. Реакции электрофильного замещения
Реакции замещения аренов протекают иными путями и в иных условиях, чем у предельных углеводородов. Алканы вступают в реакции замещения в основном по радикальному механизму. Наличие у бензольного кольца подвижного
-электронного облака, его пространственная доступность создает предпосылки для протекания реакций электрофильного замещения. В этом случае молекула реагента должна иметь атом с дефицитом электронной плотности, чему в большинстве случаев способствует использование особого типа катализаторов. Механизм реакции электрофильного замещения аренов можно представить следующим образом:
Х

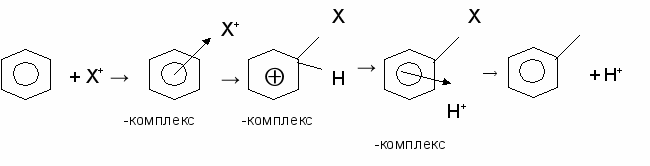
Электрофильный реагент Х+, обладающий дефицитом электронной плотности или положительным зарядом, взаимодействует с
-электронным облаком и образует с молекулой арена -комплекс. Затем ароматическая система разрушается, два из шести -электронов образуют  -связь между частицей Х+ и одним из атомов углерода цикла. Такое промежуточное состояние называется -комплекс. Поскольку ароматическое сопряжение является энергетически выгодным, для его восстановления -комплекс постепенно вытесняет протон, в результате чего вновь образуется -комплекс, который далее превращается в продукты реакции.
-связь между частицей Х+ и одним из атомов углерода цикла. Такое промежуточное состояние называется -комплекс. Поскольку ароматическое сопряжение является энергетически выгодным, для его восстановления -комплекс постепенно вытесняет протон, в результате чего вновь образуется -комплекс, который далее превращается в продукты реакции.Важнейшими реакциями электрофильного замещения Аренов являются галогенирование, алкилирование, нитрование, сульфирование.
1. Галогенирование
При взаимодействии бензола с хлором и бромом образуются моногалогензамещенные производные:
 CI
CI
+ CI2
 + HCI
+ HCIРеакция протекает в присутствии катализаторов – безводных галогенидов алюминия, железа (III) или цинка, называемых катализаторами Фриделя – Крафтса. Роль катализатора сводится к поляризации неполярной связи CI-CI с образованием электрофильной частицы, атакующей электронную плотность бензольного кольца:
CI-CI + FeCI3 → CI+[FeCI4]-
2. Алкилирование
Замещение атома водорода в бензольном кольце на алкильный радикал осуществляется под действием галогеналканов в присутствии катализатора Фриделя-Крафтса:
CH3
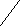
 + HCI
+ HCI 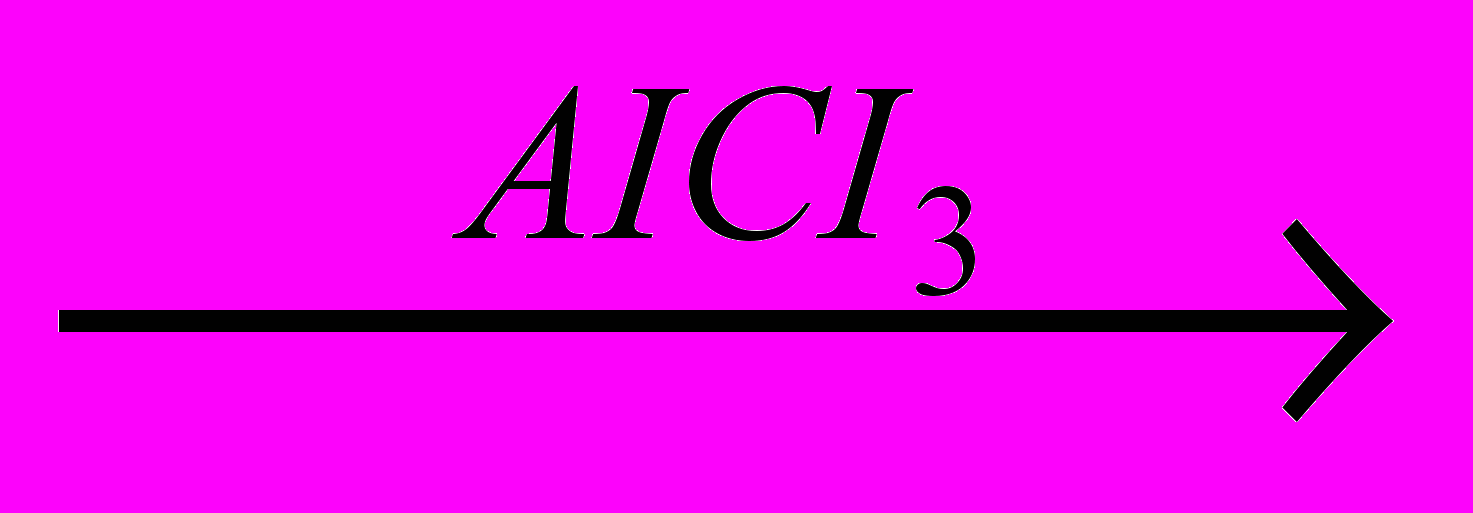 + HCI
+ HCI Еще одним удобным алкилирующим реагентом являются алкены. Реакция осуществляется в присутствии катализатора AI2O3. Алкилирование бензола этиленом приводит к образованию этилбензола, используемого для получения стирола и далее – бутадиен-стирольных каучуков. Если в качестве реагента взять пропен, то в результате реакции образуется еще один важный полупродукт органического синтеза – изопропилбензол (кумол), из которого получают фенол и ацетон:
 CH-CH3 + CH2=CH-CH3
CH-CH3 + CH2=CH-CH3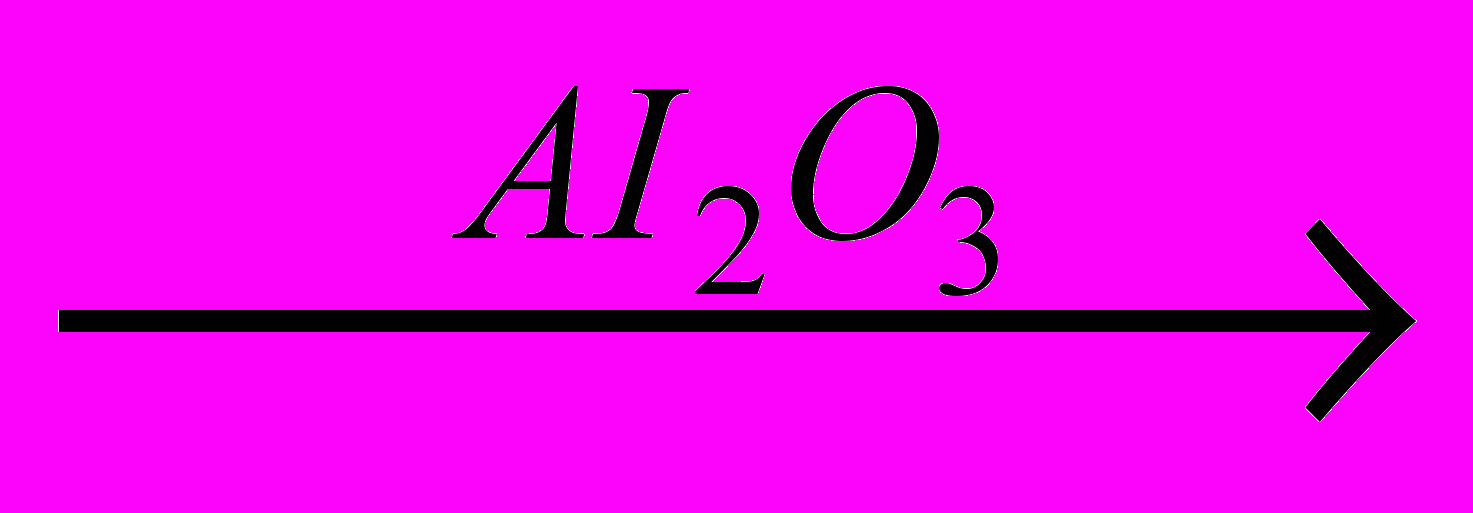 CH3
CH33. Нитрование
Замещение атома водорода в бензольном кольце происходит под действием смеси концентрированных азотной и серной кислот, называемой нитрирующей смесью:
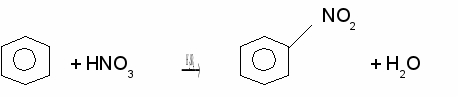
Нитробензол
Роль концентрированной серной кислоты сводится к генерированию электрофильной частицы – так называемого катиона нитрования:
H2SO4 + HO-NO2 → HSO
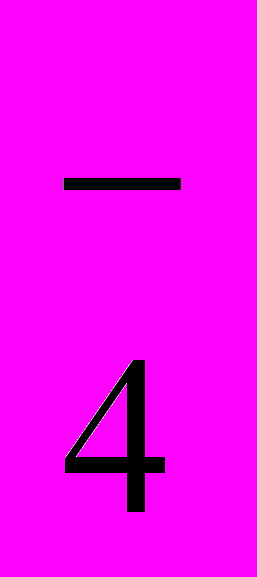 + NO
+ NO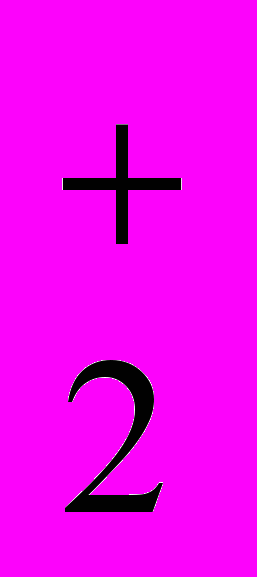 + H2O
+ H2O4. Сульфирование
Под действием концентрированной серной кислоты происходит замещение атома водорода на сульфогруппу –SO3H, представляющую собой остаток серной кислоты без одной гидроксогруппы:
 SO3H + HO – SO3H ↔ + H2O
SO3H + HO – SO3H ↔ + H2Oбензосульфокислота
Следует отметить, что реакция сульфирования, в отличие от других реакций электрофильного замещения, является обратимой.
2. Реакции присоединения
Разрушение ароматической системы с помощью реакций присоединения протекает с трудом, в жестких условиях.
1. Гидрирование
Присоединение водорода к бензолу и его гомологам происходит при повышенной температуре, давлении, в присутствии катализаторов гидрирования (Pt, Pd, Ni):
+ 3Н2 
2. Радикальное галогенирование
H CI
При облучении УФ-светом смеси паров бензола с хлором происходит присоединение трех молекул галогена с образованием гексахлорциклогексана (гексахлорана), применяемого в сельском хозяйстве для борьбы с саранчой и клещами:

H
CI
CI
H
HCI
CI
H
+ 3CI3

CI H
Такая реакция характерна только для бензола. Его гомологи в условиях радикального замещения галогенируются только по
 -углеродному атому (т. Е. ближнему к кольцу) боковой цепи независимо от его длины: СН2-СН3 CHCI-CH3
-углеродному атому (т. Е. ближнему к кольцу) боковой цепи независимо от его длины: СН2-СН3 CHCI-CH3+ CI2
+ HCIВ случае достаточного количества хлора замещению подвергается второй атом водорода в
-положении. 3.Реакции окисления
Ароматическое кольцо устойчиво к действию окислителей. Бензол при обычных условиях не окисляется кислородом воздуха, не обесцвечивает водный раствор перманганата калия.
1. Горение
Бензол и его гомологи горят на воздухе коптящим пламенем:
2С6Н6 + 15О2 → 12СО2 + 6Н2О
2. Окисление алкилбензолов
Гомологи бензола окисляются при нагревании водным раствором перманганата калия. Ароматическое кольцо при этом не затрагивается, а окисление идет по
-углеродному атому боковой цепи. Независимо от ее длины в качестве основного продукта образуется бензойная кислота: CH3 СООН СН2СН2СН3
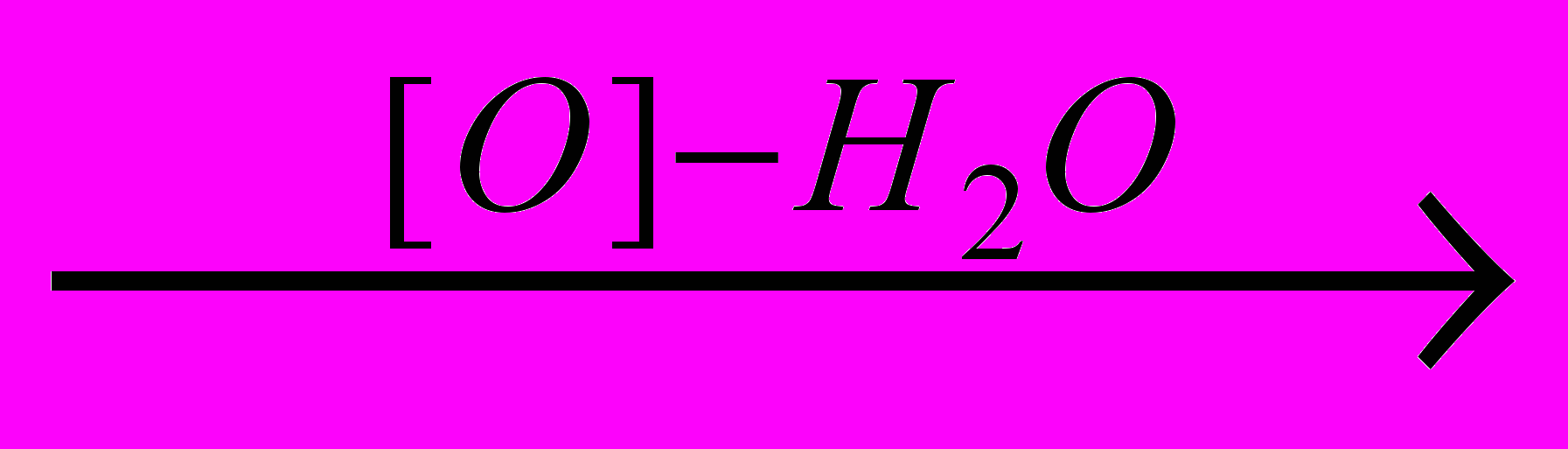
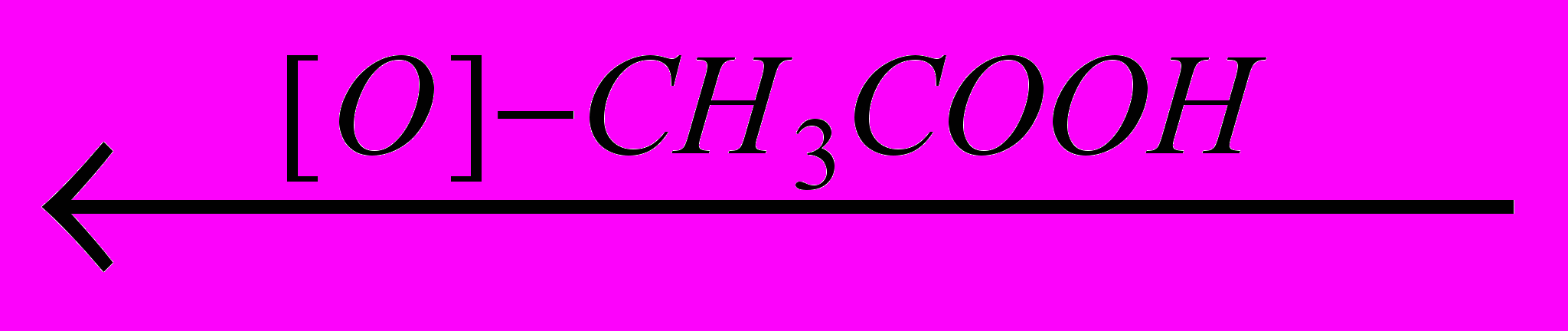
2. Способы получения ароматических углеводородов
Важнейшими природными источниками ароматических углеводородов являются нефть, каменный уголь (продукты его переработки), а также газы, образующиеся при коксовании угля.
Важнейшими синтетическими способами получения бензола и его гомологов являются следующие.
1. Ароматизация алканов и циклоалканов
Основным промышленным способом получения Аренов является дегидрирование углеводородов нефти. Предельные углеводороды, начиная с гексана и далее, а также циклогексан и его алкилзамещенные производные при пропускании над нагретым до температуры 3000С платиновым катализатором дегидрируются, причем для углеводородов с открытой цепью происходит замыкание шестичленного цикла:
СН3 CH3-CH2-CH2-CH2-CH2-CH2-CH3  + 4Н2
+ 4Н2  + Н2
+ Н2 В качестве катализатора можно использовать более дешевый оксид хрома (III). Таким способом получают низшие представители аренов: бензол, толуол, ксилолы.
2. Тримеризация ацетилена
При пропускании ацетилена над раскаленным углем образуется бензол (реакция Зелинского):
3CH≡CH 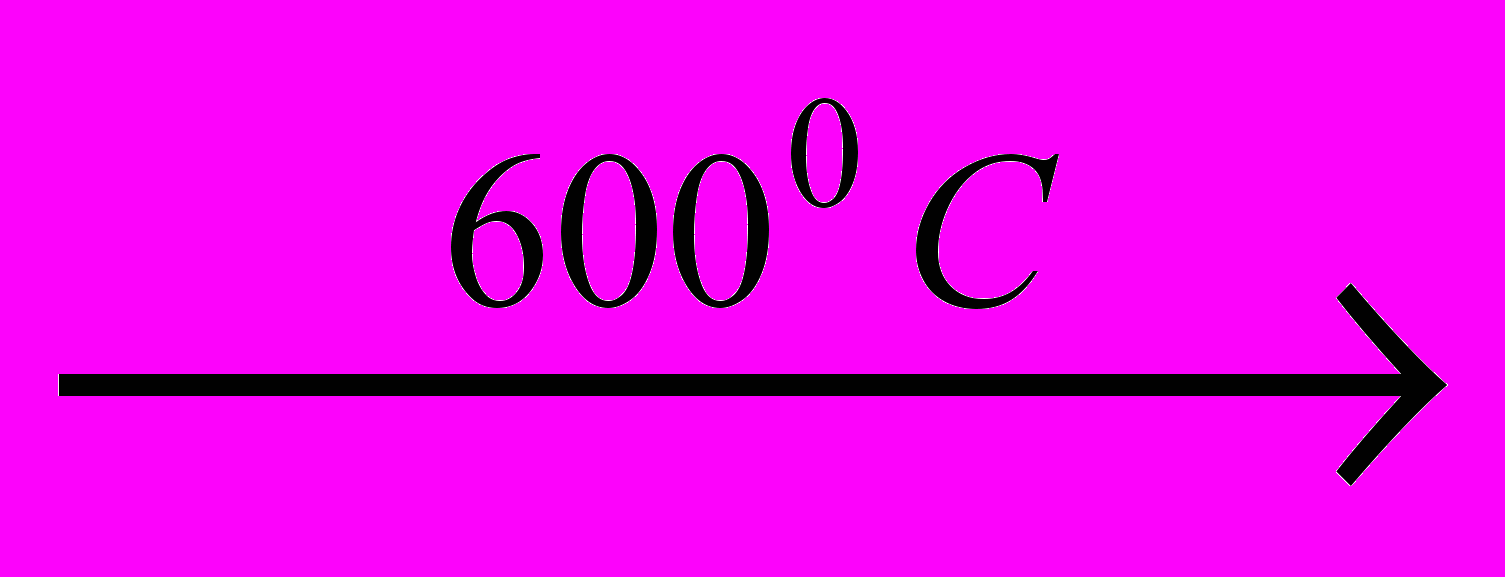
Промышленного значения данный способ не имеет, однако он важен для понимания генетической связи между углеводородами различных типов.
3. Алкилирование бензола
Гомологи бензола получают его взаимодействием с галогеналканами или алкенами в присутствии катализаторов:
R + RCI + HCI4. Пиролиз солей ароматических кислот
При сплавлении солей ароматических карбоновых кислот с щелочами образуются ароматические углеводороды:
C6H5COOK + KOH
 C6H6↑ + K2CO3
C6H6↑ + K2CO3 Эта реакция аналогична получению алканов пиролизом солей предельных карбоновых кислот.
3. Сравнение методов получения ароматических углеводородов
Изомеризация ароматических углеводородов
В ксилольных фракциях, выделенных из продуктов переработки каменного угля или нефти, содержится сравнительно мало наиболее ценных изомеров - п- и о-ксилола. В связи с этим для дополнительного получения этих углеводородов разработаны и нашли промышленное применение методы изомеризации гомологов бензола.
Изомеризация гомологов бензола является обратимым процессом, причем для ксилолов устанавливается равновесие между всеми тремя изомерами:
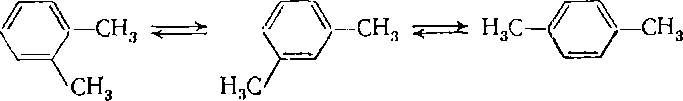
В равновесной смеси при умеренной температуре (25 - 75 °С) содержится - 60% мета, 24% пара и 16% орто-ксилола, а при 400 - 500°С - 52% мета, 23% пара и 25% орто-ксилола. Такое высокое содержание мета-изомера указывает на его более высокую термодинамическую стабильность.
Изомеризация происходит под влиянием катализаторов кислотного типа. Среди них наиболее активен хлористый алюминий, способный вызывать реакцию в жидкой фазе уже при 50 °С. В присутствии гетерогенного алюмосиликатного катализатора требуется более высокая температура (400 - 500°С), и процесс проводят в газовой фазе. В последнее время получили распространение и цеолитные катализаторы. В любом случае реакция протекает через промежуточное образование
-комплексов, образующихся из ароматического соединения и протона, генерируемого катализатором. Дальнейший процесс состоит в образовании -комплекса и миграции метальной группы к соседнему углеродному атому с последующей отдачей протона: 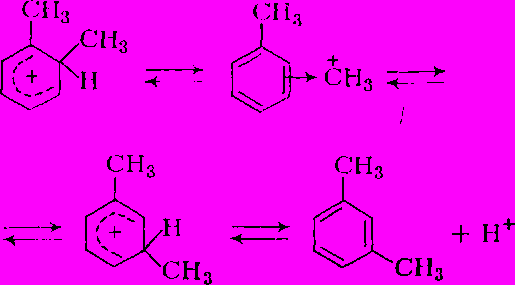
При изомеризации алкилароматических углеводородов всегда присутствует небольшое количество побочных продуктов, представляющих собой замещенные бензолы меньшей и большей степени алкилирования по сравнению с исходным соединением. Так, при переработке ксилолов получается около 3% низших гомологов (бензол и толуол) и 2- 3% полиметилбензолов.
В промышленности процесс изомеризации ксилолов всегда комбинируют с установками разделения соответствующих фракций.
Другой процесс, подобный изомеризации, состоит в диспропорционировании метилбензолов с теми же катализаторами, но в более жестких условиях. Он нашел применение для превращения избыточного толуола в бензол и ксилолы, причем реакция также протекает через
-комплекс и сопровождается межмолекулярной миграцией метальной группы:C6H6 + C6H4(CH3)2 + H+
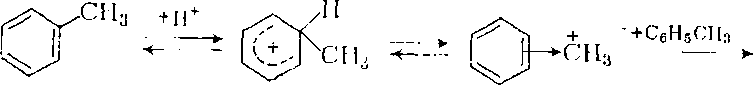
Этот процесс проводят в газовой фазе на алюмоcиликатном или цеолитном катализаторе при 350 – 530°С и 1- 1,2 МПа в присутствии водорода, который препятствует коксообразованию и удлиняет срок службы катализатора.
Деалкилирование гомологов бензола и нафталина
Большие масштабы потребления бензола и нафталина наряду с наличием избыточных количеств толуола и метилнафталинов обусловили практическое значение процессов деалкилирования (деметилирование) ароматических углеводородов. В настоящее время этим путем получают значительное количество бензола и нафталина.
Деалкилирование ароматических углеводородов основано на их деструктивной гидрогенизации (гидрогенолиз) с расщеплением углерод-углеродной связи между ароматическим ядром и алкильной группой:
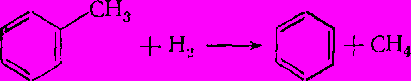
Эту реакцию можно осуществлять без катализаторов (термическое деалкилирование) или с гетерогенными контактами (каталитическое деалкилирование).
Термическое деалкилирование толуола успешно протекает при 700 – 760°С. Во избежание чрезмерного образования кокса и снижения выхода целевых продуктов процесс необходимо проводить при 4 - 5 МПа и избытке водорода (4:1) по отношению к толуолу. В этих условиях предотвращаются реакции дегидроконденсации ароматических соединений, являющиеся причиной коксообразования. Сообщается о ≈ 50% степени конверсии толуола с последующей рециркуляцией непрореагировавшего сырья, когда выход бензола составляет до 98% от теоретического.
Каталитическое деалкилирование ароматических углеводородов осуществляется с такими дегидро-гидрирующими катализаторами, которые активны в отношении деструктивного гидрирования и не затрагивают ароматическое ядро. К ним относятся оксиды молибдена, кобальта и хрома, причем практическое применение нашел оксид хрома, нанесенный на активный оксид алюминия. Во избежание отложения кокса па поверхности катализаторов и быстрого его дезактивирования проводят процесс при давлении водорода 3 - 10 МПа. Водород, как и при термическом деалкилировании и каталитическом риформинге, препятствует реакциям дегидроконденсации. Температура при каталитическом деалкилировании лишь немного ниже, чем при термическом, и составляет 580 – 620°С. Выход бензола из толуола может превышать 95% от теоретического.
4. Технология процесса получения ароматических углеводородов
Схема комплексной переработки ксилольной фракции изображена на рис. 1.
В первой ректификационной колонне 1 из исходной ксилольной фракции отгоняется более летучий этилбензол. Во второй колонне 2 проводится совместная ректификация вновь поступающих и изомеризованных ксилолов. В кубе этой колонны собирают наименее летучий о-ксилол, который выводят из системы в виде готового продукта. Смесь мета- и пара-ксилолов, выходящих из верхней части колонны 2, направляют на установку 4 первой ступени кристаллизации, где охлаждают смесь до минус 50 - минус 70 °С. Выпавшие кристаллы отделяют центрифугированием. Маточный раствор, полученный при фильтровании, содержит 75 - 85% м-ксилола. Его направляют на установку 6 для изомеризации; при этом образуется дополнительное количество орто- и пара-ксилолов. Из изомеризованного продукта вначале отделяют ректификацией в колонне 3 побочные продукты (бензол, толуол и полиметилбензолы), а ксилолы направляют в колонну 2. Таким образом, значительная часть продукта циркулирует в стадиях 2 – 4 – 6 - 2.
Твердый продукт, полученный при первой кристаллизации, содержит только 70 - 80% пара-ксилола. Его расплавляют и подвергают повторной кристаллизации на установке 5; после центрифугирования выделяют 98% пара-ксилол. Маточный раствор после второй кристаллизации содержит значительное количество п-изомера, поэтому его возвращают на первую стадию кристаллизации.
При переработке ксилольной фракции с целью получения только одного изомера, например п-ксилола, отпадает необходимость во второй ректификационной колонне, а остальные стадии процесса остаются теми же. Таким путем удается получить 70% п-ксилола (считая на исходную ксилольную фракцию).
Кроме кристаллизации, для выделения п-ксилола применяют Парекс-процесс адсорбции цеолитами.




Этилбензол



4

С6 – С7



1
2
5

3




Ксилольная фракция
п-Ксилол

С6
6






о-Ксилол
С9
Рис. 1. Технологическая схема разделения ксилольной фракции, совмещенного с изомеризацией
1. Колонна отгонки этилбензола, 2. Колонна выделения о-ксилола, 3. Колонна отделения легкой и тяжелой фракций, 4. Установка первой ступени кристаллизации, 5. Установка второй ступени кристаллизации, 6. Установка изомеризации
5. Оценка методов получения ароматических углеводородов
При коксовании, пиролизе и риформинге получают следующие выходы ароматических углеводородов (в кг на 1 т ископаемого сырья) приведенные в таблице 1:
Таблица 1
Выход ароматических углеводородов
| Наименование | Коксование угля | Пиролиз лигроина | Платформинг лигроина |
| 1 | 2 | 3 | 4 |
| Бензол | 6 – 6,5 | 7 - 9 | 5 – 10 |
| Толуол | 1,5 | 4 - 6 | 5 - 7 |
| Ксилолы | 0,3 | 1,5 – 2,5 | 1 – 3 |
| Нафталин | 2 – 2,5 | 2 – 3 | - |
При расчете на исходную нефтяную фракцию выход ароматических углеводородов оказывается примерно в 10 раз выше указанного.
При оценке роли каждого процесса следует иметь в виду, что при коксовании и пиролизе ароматические углеводороды получаются как побочные вещества при получении кокса и олефинов, и выделение повышает экономическую эффективность производства. Поэтому до сих пор ≈ 10% бензольных углеводородов и весь нафталин получают коксохимическим методом. В Западной Европе преимущественное значение для производства ароматических углеводородов имеет процесс пиролиза, и его роль возрастает во всем мире в связи с переходом па жидкое сырье, дающее повышенный выход ароматических углеводородов и бутадиена. Получаемый при этом избыточный толуол выгодно перерабатывать на бензол и ксилолы (например, в США в 1976 г. 30% всего бензола производили гидродеалкилированием толуола). Наконец, только недостающее количество бензола и ксилолов целесообразно получать целевым процессом производства ароматических углеводородов - риформингом узких нефтяных фракций.
Список литературы
1. Габриэлян О. С., Остроумов И. Г. Химия. М., Дрофа, 2008;
2. Чичибабин А. Е. Основные начала органической химии. М., Госхимиздат, 1963. – 922 с.;
3. Лебедев Н. Н. Химия и технология основного органического и нефтехимического синтеза. М., Химия. 1988. – 592 с.;
4. Паушкин Я. М., Адельсон С. В., Вишнякова Т. П. Технология нефтехимического синтеза. М., 1973. – 448 с.;
5. Юкельсон И. И. Технология основного органического синтеза. М., «Химия», 1968.