
Курс Викладач Жук Л. П. Дисципліна Хімія
| Вид материала | Документы |
- Курс Викладач Жук Л. П. Дисципліна Спектральні методи дослідження органічних реагентів, 503.92kb.
- Курс Викладач Жук Л. П. Дисципліна Методологія та організація наукових досліджень, 878.91kb.
- Курс Викладач Безуглий В. В дисципліна Економічна та соціальна географія України модуль, 2428.67kb.
- Курс I хімія Викладач доцент Шевченко Л. В. Лекція на тему, 627.62kb.
- Курс 1, 2 на базі повної загальної середньої освіти форма навчання, 558.98kb.
- Курс: 1 Статус дисципліни: обов’язкова Стаціонарне навчання Години на тиждень Триместр, 432.82kb.
- Робоча програма навчальної дисципліни екоаналітична хімія напряму підготовки 040101, 154.06kb.
- Робоча програма навчальної дисципліни математичне моделювання аналітичних систем напряму, 172.48kb.
- А. І. Жук 2008 г, 802.37kb.
- Дисципліна «фінанси» Викладач: доц. Румик І.І. Завдання для самостійної роботи студентів, 273.36kb.
Факультет Геолого-географічний
Спеціальність Гідрогеологія
1курс
Викладач Жук Л.П.
Дисципліна Хімія
Розчини. Теорія електролітичної дисоціації.
Розчини мають величезне значення у природі, промисловості, медицині. Рослини засвоюють речовини у вигляді розчинів. Засвоєння їжі пов’язане з переводом поживних речовин у розчин. Усі природні води є розчинами. Розчинами є найважливіші фізіологічні рідини – плазма, кров. Спинномозкова рідина, лімфа. Більшість хімічних реакцій відбувається у розчинах.
Отже, розчини – це гомогенні (однорідні) системи, що складаються з двох і більше компонентів і продуктів їх взаємодії.
За агрегатним станом розчини бувають рідкі, тверді і газоподібні. Прикладом рідких розчинів можуть бути розчини солей у воді, прикладом твердих – сплав нікелю й міді (з яких виготовляють монети) або сплав срібла й золота, прикладом газоподібних – суміші газів, повітря. Найбільше значення мають рідкі, а особливо – водні, розчини.
Будь-який розчин складається з розчинених речовин і розчинника, хоч ці поняття до певної міри умовні. Наприклад, залежно від співвідношення кількостей спирту й води ця система може бути розчином спирту у воді або води у спирті. Як правило, розчинником вважають той компонент, який у розчині знаходиться у тому ж агрегатному стані, що й до розчинення.
Самим поширеним розчинником на нашій планеті є вода. Тіло середньої людини масою 70 кг містить приблизно 40 кг води. У тварин і рослинних організмів вода складає від 50 до 90-95 %.
Внаслідок своїх аномальних властивостей вода – універсальний розчинник, чудово приладнаний для життєдіяльності.
Насамперед вода добре розчиняє іонні й більшість полярних сполук. Така властивість води пов’язана значною мірою з її високою діелектричною проникністю (ε = 78,5). Оскільки сили тяжіння між іонами, згідно закону Кулона, змінюються обернено пропорційно величині ε, тяжіння між іонами зменшується приблизно у 80 разів при розчиненні іонних сполук у воді. Внаслідок більшість іонних сполук дисоціюють і відрізняються високою розчинністю у воді.
Інший багато чисельний клас речовин, добре розчинних у воді, включає такі полярні органічні сполуки, як сахара, альдегіди, кетони, спирти. Їх розчинність у воді пояснюється здатністю молекул води до утворення полярних зв’язків з полярними функціональними групами цих речовин, наприклад, з гідроксильними групами спиртів і сахарів або з атомом оксигену карбонільної групи альдегідів і кетонів.
Внаслідок високої полярності вода викликає гідроліз речовин.
Оскільки вода складає основну частину внутрішнього середовища організмів, то вона забезпечує процеси всмоктування, пересування поживних речовин і продуктів обміну. Необхідно відзначити, що вода є кінцевим продуктом біологічного окислення речовин, яке супроводжується виділенням великої кількості енергії – приблизно 21 – 29 кДж/моль .
Важливі й інші аномальні властивості води: високий поверхневий натяг, низька в’язкість, високі температури плавлення й кипіння й більш висока густина у рідкому стані, аніж у твердому.
Молекула води утворюється з двох атомів гідрогену й атома оксигену. Кут між ними складає 104,5 0. Внаслідок асиметрії у розподілі електронів навколо атома оксигену центр негативного електричного заряду (неподіленої пари електронів) електронної хмари не співпадає з центром позитивного заряду атома оксигену. Це призводить до появи значного електричного дипольного моменту молекул води, який і визначає її полярні властивості й добру розчинність полярних і низьку розчинність неполярних речовин у воді.
Для води характерна наявність асоціатів – груп молекул, з’єднаних водневими зв’язками.
Залежно від спорідненості до води функціональні групи ділять на гідрофільні (що притягають воду), які легко сольватуються водою, гідрофобні (що відштовхують воду) й дифільні.
До гідрофільних груп відносяться полярні функціональні групи: гідроксильна –ОН, аміно – NH2, тіольна – SH, карбоксильна – СООН. До гідрофобних – неполярні групи, наприклад вуглеводневі радікали: СН3(СН2)п -, С6Н5 -. До дифільних відносять речовини (амінокислоти, білки, нуклеїнові кислоти), молекули яких містять як гідрофільні групи, так і гідрофобні.
Класифікація розчинів.
Розчини речовин з молярною масою менше 5000 г/моль називають розчинами низькомолекулярних сполук (НМС), а розчини речовин з молярною масою понад 5000 г/моль – розчинами високомолекулярних сполук (ВМС).
За наявністю або відсутністю електролітичної дисоціації розчини НМС діляться на розчини електролітів і неелектролітів.
Речовини, що розпадаються на іони в розчинах або розплавах і тому проводять електричний струм – електроліти (до них належать кислоти, основи і майже усі солі). Речовини, які за таких самих умов не розпадаються на іони і електричний струм не проводять – неелектроліти (до них належать більшість органічних сполук, а також речовини, в молекулах яких є тільки ковалентні неполярні або мало полярні зв’язки).
Розчини можуть бути насиченими, ненасиченими і пересиченими. Насиченим називається розчин, який перебуває у динамічній рівновазі з надлишком розчиненої речовини. У насиченому розчині за даної температури міститься максимально можлива кількість розчиненої речовини. У ненасиченому розчині міститься менше речовини, а в пересиченому – більше, ніж у насиченому. Пересичені розчини досить нестійкі. Легке струшування посудини або добавляння в розчин кристала солі викликає випадання в осад надлишку розчиненої речовини.
Розчини НМС – електролітів і неелектролітів – називають істиними на відміну від колоїдних розчинів. Істині розчини характеризуються гомогенністю складу й відсутністю поверхні розподілу між розчиненою речовиною й розчинником. Розмір розчинених часток – іонів і молекул – менше як 10-9 м.
Важливою характеристикою будь-якого розчину є його концентрація. Цією величиною визначається більшість властивостей розчинів.
Концентрація речовини – це величина, що вимірюється кількістю розчиненої речовини, що міститься у певній масі чи об’ємі розчину або розчинника.
Існують різні способи чисельного вираження складу розчинів.
1. Масова доля ω - кількість г речовини, що міститься у 100 г розчину чи сухої речовини, виражається у долях одиниці або відсотках %:
ω (Х) = m (Х)/m (розчину)
ω (Х) = m (Х)*100 %/m (розчину).
2. Молярна концентрація c (Х) – кількість молей речовини, що міститься у 1 л (або дм3) розчину, виражається у моль/л, моль/дм3:
c (Х) = n (Х)/ V (розчину) = m (Х)/ M (Х)V (розчину).
В хімії широко користуються поняттям еквіваленту та фактору еквівалентності.
Еквівалентом називають реальну або умовну частку речовини Х, яка може приєднувати, вивільнювати або бути будь-яким іншим чином еквівалентна одному іону гідрогену в даній кислотно-основній реакції або одному електрону в окислювально-відновній реакції.
Під умовною часткою розуміють як реально існуючі частинки (молекули, іони, електрони), так і долі таких частинок (наприклад, ½ іону) або їх групи.
Кількість речовини еквіваленту є моль.
Фактор еквівалентності f екв.(Х) – це число, яке позначає, яка доля реальної частки речовини Х еквівалентна одному іону гідрогену в даній кислотно-основній реакції або одному електрону в окислювально-відновній реакції.
Фактор еквівалентності розраховують на основі стехіометрії даної реакції з рівності
f екв.(Х) = 1/z, де z – основність кислоти або кислотність основи даної кислотно-основної реакції, а також електронів, що приймають участь у даній окислювально-відновній реакції. z- величина безрозмірна.
Молярна маса еквівалента речовини Х (маса одного моля еквівалента речовини) – величина, яка вимірюється добутком фактора еквівалентності на молярну масу речовини Х:
M (1/z X) = 1/z M (X), де M (1/z X) – молярна маса еквівалента.
Одиниці вимірювання молярної маси еквівалента – г/моль.
3. Молярна концентрація еквівалентів (нормальна концентрація) c(1/zХ). – кількість молей еквівалентів речовини, що міститься у 1 л (або дм3) розчину, виражається у моль/л, моль/дм3:
c (1/z X) = n (1/z X)/ V(розчину), де n (1/z X) – кількість речовини еквівалентів, моль.
Молярна концентрація і молярна концентрація еквівалентів зв’язані співвідношенням:
c (1/z (X)) = zc (X).
4. Титр розчину T (Х) – концентрація розчину, яка показує кількість розчиненої речовини у г, що міститься у 1 мл розчину:
T (Х) = m (X)/V (розчину).
Теорія електролітичної дисоціації.
Отже пригадаємо, що електролітична дисоціація – це розпад електролітів на іони під час розчинення їх у воді.
Для пояснення особливостей водних розчинів електролітів шведський учений С. Арреніус у 1887 р. запропонував теорію електролітичної дисоціації. Сучасний зміст цієї теорій можна звести до трьох положень:
1. Електроліти під час розчинення у воді розпадаються (дисоціюють) на іони – позитивні і негативні.
Іони перебувають у стійкіших електронних станах, ніж атоми. Вони можуть складатися з одного атома – прості іони (Na+, Mg2+, Al3+) або з кількох атомів – складні іони (NO3-, SO42-, PO43-).
Сама назва “іон” – у перекладі з грецької означає “мандрівний”. У розчині іони безладно переміщуються (“мандрують”) у різних напрямках.
2. Під дією електричного струму іони набувають напрямленого руху: позитивно заряджені іони переміщуються до катода, негативно заряджені – до анода.. Тому перші називаються катіонами, а другі – аніонами.
3. Дисоціація – оборотний процес: паралельно з розщепленням молекул на іони (дисоціація) відбувається процес сполучення іонів (асоціація).
Тому в рівняннях електролітичної дисоціації замість знака “дорівнює” ставлять знак оборотності.
Теорія електролітичної дисоціації є однією з основних теорій в неорганічній хімії і повністю узгоджується з атомно-молекулярним вченням і теорією будови атома.
В роботах І.А.Каблукова і В.А.Кістяковського була розвинена теорія електролітичної дисоціації на основі хімічної (гідратної) теорії розчинення Д.І.Менделєєва. Вони відзначали. Що електролітична дисоціація викликається не тільки послабленням тяжіння іонів у розчиннику, але й сольватацією – взаємодією полярних молекул розчинника з частками розчиненої речовини. Саме сольватація є головна причина дисоціації частинок розчиненої речовини.
При розчиненні електролітів у воді диполі води (полярного розчинника) за рахунок орієнтаційної диполь-діпольної або іон-діпольної взаємодії притягуються до полярних молекул або іонів речовини, що розчиняється. Таким чином, першою стадією дисоціації завжди є гідратація (сольватація). У речовинах з полярними молекулами, наприклад у хлороводні HCl, під дією молекул розчинника відбувається сильне зміщення зв’язуючи електронів і зв’язок H – Cl стає іонним. Цю другу стадію дисоціації називають поляризацією молекул речовини, що розчиняється. Третьою стадією є саме дисоціація, тобто руйнування поляризованої молекули й утворення гідратованих іонів.
Роль розчинника полягає не тільки у створенні умов для поляризації й розділення іонів протилежного знака, але і в уповільненні їх рекомбінації. Іони, що перейшли у розчин, соль ватовані (гідратовані), тобто внаслідок дисоціації утворюються не вільні іони, а відносно стійкі сполуки іонів з молекулами розчинника – сольвати іонів (якщо розчинник вода – гідрати іонів). Число молекул розчинника в сольватній оболонці іонів залежить від природи іонів, температури і концентрації розчину. Тому до рівняння дисоціації, як правило, пишуть формули іонів, а не їх гідратів і сольватів. Іноді роблять виключення для іона Н+, аби підкреслити хімічний характер взаємодії протона з молекулою води з утворенням стійкого оксоній-іона:
Н+ + Н2О = Н3О+.
Сольватні оболонки послаблюють електростатичну взаємодію іонів згідно закону Кулона тим сильніше, чим більша діелектрична проникненість ε розчинника. Тому електролітична дисоціація можлива не тільки в воді (ε = 81), але і в HCN (ε = 95), етанолі (ε = 27), мурашиній кислоті (ε = 58) та інших розчинниках.
Ступінь дисоціації (іонізації). Сила електролітів.
Розрізняють сильні і слабі електроліти. Електроліти, які практично повністю дисоціюють на іони – сильні, а які не повністю іонізуються – слабі.
У розчинах слабих електролітів поряд з іонами існують неіонізовані молекули. Для кількісної характеристики повноти дисоціації введене поняття ступеня дисоціації.
Ступінь дисоціації електроліта – це відношення числа молекул, що розпалися на іони, до загального числа молекул розчиненої речовини.
Інакше кажучи,αі – доля молекул електроліта, що розпалися на іони:
α і = n/N.
За ступенем дисоціації електроліти умовно поділяють на сильні (αі > 30%) і слабі (αі < 3 %). У проміжку 3 % < α і < 30 % електроліти середньої сили.
На ступінь дисоціації впливає:
- температура,
- концентрація,
- додавання однойменних іонів.
Кількісно електролітичну дисоціацію як рівноважний оборотній процес можна охарактеризувати константою дисоціації (іонізації), що визначається законом діючих мас.
Закон діючих мас, точно говорячи, застосовується лише до оборотних реакцій, тобто до розчинів слабких електролітів. Якщо дисоціацію електроліта уявити як рівноважний процес:
KtnAnm↔ nKtm+ + mAnn-.
Відповідно до закона діючих мас константу рівноваги запишемо так:
Kд = [Ktm+]n[Ann-]m / [KtnAnm].
Це рівняння справедливе лише для розбавлених розчинів слабких електролітів. Чим більша константа дисоціації Кд, тим сильніший електроліт.
На відміну від ступеня дисоціації Кд залежить тільки від природи електроліта і температури, але не залежить від концентрації розчину! Таким чином, і константа дисоціації Кд, і ступінь електролітичної дисоціації αі – кількісні характеристики дисоціації. Зрозуміло, що між ними існує зв’язок.
Так для слабкого бінарного електроліта концентрація [Kt+] = [An-] = с(Х)αі, а концентрація недисоційованих молекул [KtAn] = с (Х) – с (Х)*αі = = с (Х) (1 – αі).
Підставивши ці значення у вираз для константи дисоціації, одержимо:
Кд = с (Х)*αі*с (Х)*αі / с (Х)*(1 - αі) = αі2*с (Х)/ (1 - αі).
Це співвідношення називають законом розведення Оствальда (1888).
Для слабкого електроліта αі << 1, тоді величиною αі у знаменнику можна знехтувати і рівняння прийме вигляд:
Кд ≈ αі2*с (Х) або αі ≈ √Кд/с (Х).
Якщо замість 1/с (Х) підставити V(Х) = 1/с (Х), яке називається розведенням, рівняння набуде вигляду
αі ≈ √КдV (Х).
Відповідно закон Оствальда може бути сформульований так:
ступінь дисоціації слабкого електроліту зростає з розведенням розчину.
Сильні електроліти не підкоряються цьому закону.
Багатоосновні кислоти і багатокислотні основи дисоціюють ступінчасто. Дисоціація за ступенями характеризується ступінчастими константами. На прикладі фосфорної кислоти.
Сумарне рівняння. Сумарна константа дисоціації Ксум = К1К2К3. При цьому К1 > К2 > К3. Замість константи дисоціації зручніше користуватися її десятковим логарифмом, взятим з оберненим знаком : рКд = - lg Кд. Наприклад для оцтової кислоти Кд = 1,76*10-5, і відповідно рКд = 4,76.
Теорія розчинів сильних електролітів Дебая і Хюккеля.
У водних розчинах сильні електроліти практично повністю дисоційовані. І на відміну від розчинів слабких електролітів їх розчини містять значно більше число іонів. В не дуже розбавлених розчинах відстань між іонами мала. Це приводить до сильної міжіонної взаємодії. Внаслідок біля кожного іона знаходяться переважно іони протилежного знаку: утворюються іонні пари Kt+An-, триплети Kt+An-Kt+ або An-Kt+An- і “іонні атмосфери”. Як наслідок, виникає ефект зменшення числа іонів, що приймають участь у хімічних процесах.
При руханні іонів під дією електричного поля іон даного знака рухається до протилежно зарядженого електрода, а оточуюча його атмосфера гальмує рух і зменшує рухливість іона. Чим більша концентрація розчину, тим більше проявляється гальмівна дія “іонної атмосфери”. Крім того, рух іонів гальмується також сольватними оболонками. При розведенні розчину вплив “іонної атмосфери” зменшується, а при безкінечному розведенні зникає, адже іони практично не взаємодіють між собою.
Таким чином, можна вважати, що в усіх процесах у розчинах електролітів приймають участь лише “активні іони”, тобто іони, які не приймають участі в даний момент в між іонних взаємодіях. У зв’язку з цим для оцінки концентраційних ефектів у розчинах сильних електролітів вводиться величина, яка називається активністю α:
Під активністю електроліта Х розуміють ефективну концентрацію, у відповідності до якої він приймає участь у різних процесах.
Активність зв’язана з істиною концентрацією розчиненої речовини співвідношенням:
α (Х) = f (Х)*с (Х),
де α (Х) – активність електроліта, моль/л; с (Х) – його концентрація, моль/л; f (Х) – коефіцієнт активності (величина безрозмірна).
Коефіцієнт активності f (Х) виражає відхилення розчину з концентрацією с (Х) від поведінки розчину при безкінечному розведенні, тобто за відсутності між іонних взаємодій.
Іонна сила розчину. В розведених розчинах природа іонів незначно впливає на значення коефіцієнтів активності, оскільки між іонні взаємодії визначаються лише зарядами іонів та їх концентрацією. При цьому кількісною характеристикою між іонних електростатичних взаємодій є іонна сила розчину І:
Іонною силою розчину називають величину, що вимірюється напівсумою добутку концентрацій усіх іонів, що знаходяться у розчині, на квадрат їх заряду.
І = ½ ∑(сіzі2),
де І – іонна сила розчину; сі – молярні концентрації іонів; zі – заряди іонів.
П. Дебай і Е. Хюккель в 1923 р. показали, що для розбавлених розчинів з іонною силою І≤ 0,01 коефіцієнти активності можна розрахувати за формулою:
lg fi = - 0,5 zi2√I,
де fi – коефіцієнт активності іона, а zi – його заряд.
Якщо користуватися значеннями активностей замість концентрацій, то закон діючих мас можна застосовувати до сильних електролітів і концентрованих розчинів слабких електролітів. Для таких розчинів константу рівноваги слід виражати через активності.
Теорії кислот і основ.
Більшість електролітів, наприклад гідроксиди різних елементів Е, проявляють властивості кислот або основ. Дисоціація гідроксида ЕОН може відбуватися двома шляхами:
Е – О – Н ↔ ЕО- + Н+ (кислотний гідроксид)
Е – О – Н ↔ Е+ + НО- (основний гідроксид),
тобто розрив може відбуватися по обох зв’язкам групи Е – О – Н.
Як відомо, полярність і міцність зв’язків залежить від різниці електронегативностей елементів, розміру й ефективного заряда атомів. Якщо енергія розрива зв’язку О – Н значно менше енергії розрива зв’язку Е – О, то дисоціація відбувається по кислотному типу. Якшо ж навпаки, енергія розрива зв’язку О – Н значно більше енергії розрива енергії зв’язку Е – О, то дисоціація протікає по основному типу.
При співставимій міцності зв’язків О – Н ті Е – О дисоціація може одночасно протікати по кислотному й основному типам:
ЕО- + Н+ ↔ ЕОН ↔ Е+ + НО-
Гідроксиди такого типа називають амфолітами.
Для характеристики багатьох електролітів у водних розчинах можна використовувати поняття кислоти та основи, дані Арреніусом:
кислота – електроліт, дисоціюючий у розчинах з утворенням гідроген-іонів Н+;
основа – електроліт, дисоціюючий у розчинах з утворенням гідроксид-іонів ОН-;
амфоліт – електроліт, дисоціюючий у розчинах з утворенням як гідроген-іонів Н+, так і гідроксид-іонів ОН-.
Але ця теорія недосконала. Обмеженість можна проілюструвати таким чином.
1. Молекула NH3 не містить іона ОН-, а молекула СО2 – іона Н+, однак у водних розчинах перша проявляє властивості основи, а друга – кислоти.
2. Безводний хлороводень, який складається тільки з молекул, реагує з безводними основами.
3. Більшість електролітів, що містять гідроген, в одному розчиннику дисоціюють як кислоти, а в іншому – як основи. Наприклад. СН3СООН у воді – слабка кислота:
СН3СООН ↔ СН3СОО- +Н+
А в рідкому фтороводні – основа:
HF + СН3СООН ↔СН3СООН2+ + F-
Для пояснення таких невідповідностей були створені інші теорії кислот і основ.
Протонна теорія кислот і основ І. Бренстеда і Т. Лоурі розроблена у 1923р.
Відповідно до неї:
Кислотою називають будь-яку речовину, молекулярні частинки якого (в тому числі і іони) здатні віддавати протон, тобто бути донором протонів;
Основою називають будь-яку речовину, молекулярні частинки якого (в тому числі і іони) здатні приєднувати протон, тобто бути акцептором протонів.
Такі визначення кислот і основ дають можливість включити до їх числа не тільки молекули, але й іони. Наприклад, карбонат-іон відповідно до протонної теорії – основа, оскільки у водному розчині він приєднує протон:
СО32- + Н+ ↔ НСО3-
Відповідно до протонної теорії кислоти ділять на три типа:
1) нейтральні кислоти, наприклад HCl, H2SO4, H3PO4 та інші:
H2SO4 ↔ H+ + HSO4-
2) катіонні кислоти, які представляють собою позитивні іони, наприклад NH4+, Н3О+:
NH4+ ↔ NH3 + H+
3) аніонні кислоти, які представляють собою негативні іони, наприклад HSO4-, H2PO4-, HPO42- та інші:
HSO4- ↔ H+ + SO42-
Подібного типа класифікація є й для основ:
1) нейтральні основи, наприклад NH3, H2O, С2H5OН та інші:
NH3 + H+ ↔ NH4+
2) аніонні основи, які представляють собою негативні іони, наприклад Cl-, СН3СОО-, ОН-:
СН3СОО- + H+ ↔ СН3СООH
3) катіонні основи, які представляють собою позитивні іони, наприклад NH2 - NH3+.
Розчинники типу види, рідкого аміаку, аніони багато основних кислот, які можуть бути і донорами і акцепторами протонів, являються амфолітами.
Наприклад, в реакції
H2O + NH3 ↔ ОН- + NH4+
молекула води віддає протон і являється кислотою. Однак в реакції
H2O + HCl ↔ Н3О+ + Cl-
молекула води приєднує протон і являється основою. Отже вода – типовий амфоліт.
Процес дисоціації (іонізації) речовини відбувається в контакті з розчинником. При цьому розчинник виконує або функцію кислоти або іункцію основи. Наприклад, при розчиненні аміаку вода – кислота, а при розчиненні фтороводню – основа.
Відповідно до протонної теорії, віддаючи протон, кислота перетворюється на основу, яку називають супряженою цій кислоті:
кислота1 ↔ супряжена основа1 + Н+
тобто кожній кислоті відповідає супряжена основа. Навпаки, основа, приєднуючи протон, перетворюється на супряжену кислоту:
основа2 + Н+ ↔ супряжена кислота2
Наприклад, кислоті H2SO4 відповідає супряжена основа HSO4-, основі Cl- - супряжена кислота НCl.
Оскільки протон у розчинах не існує у вільному вигляді, кислота може віддати протон тільки основі, яка, прийнявши протон, стає кислотою. Тому відповідно до протонної теорії має місце кислотно-основна рівновага, обумовлена переносом протону:
кислота1 + основа2 ↔ кислота2 + основа1
Наприклад:
HCl + NH3 ↔ NH4+ + Cl-
кислота1 основа2 кислота2 основа1
Реакції нейтралізації, іонізації, гідролізу з точки зору протонної теорії є частковий випадок КО-рівноваги.
Реакція І типу СН3СООH + H2О ↔ СН3СОО- + Н3О+, протікаючи у прямому напрямку, представляє іонізацію оцтової кислоти, у зворотному напрямку – нейтралізацію будь-якого ацетату сильною кислотою.
Реакція ІІ типу NH4+ + H2О ↔ NH3 + Н3О+, протікаючи у прямому напрямку, представляє гідроліз будь-якої солі амонію, у зворотному напрямку – нейтралізацію аміаку сильною кислотою.
В цих кислотно-основних рівновагах вода відіграє роль основи. Але, будучи амфолітом в інших кислотно-основних рівновагах, вона може виконувати й роль кислоти, наприклад:
H2О + СН3СОО- ↔ СН3СООH + ОН-
Тепер пряма реакція кислотно-основної взаємодії представляє собою гідроліз ацетату, а зворотна – реакцію нейтралізації оцтової кислоти сильною основою.
Протеолітичні кислотно-основні рівноваги ІІІ типу можуть мати місце не тільки у воді, але і в інших розчинниках, наприклад у рідкому аміаку:
СН3СООH + NH3 ↔ СН3СОО- + NH4+
в безводному HF:
С2Н5ОH + HF ↔ С2Н5СОН2+ + F-
Дисоціація води.
Як уже було показано, вода поводе себе як амфоліт. Процес дисоціації води відповідно до теорії Бренстеда протікає за рівнянням
H2О + H2О ↔ Н3О+ + ОН-
Відбувається автоіонізація води.
Константа дисоціації води при 25 0С дорівнює:
Кд (Н2О) = а(Н+)* а(ОН-)/ а(Н2О) = 1,8*10-16 моль/л, де а(Н+), а(ОН-), а(Н2О) – активності іонів Н+, ОН- та води Н2О. Ступінь дисоціації води дуже малий, тому активності гідроген- та гідроксид-іонів в чистій воді практично дорівнюють їх концентраціям. Оскільки вода присутня у великому надлишку, її концентрація може вважатися сталою й складає 55,6 моль/л (1000 : 18 г/моль = 55,6 моль/л). Підставивши це значення до виразу для константи дисоціації, а замість активностей гідроген- та гідроксид-іонів їх концентрації, одержують новий вираз:
К (Н2О) = [Н+][ОН-] = 1*10-14 моль2/л2, або точніше К (Н2О) = а(Н+)* а(ОН-) = 1*10-14 моль2/л2. Константа К (Н2О) називається іонним добутком або константою автоіонізації (автопротолізу) води. Тобто в чистій воді або любому водному розчині при сталій температурі добуток концентрацій (активностей) гідроген- та гідроксид-іонів є величина стала, яка називається іонним добутком води.
Константа К (Н2О) залежить від температури: при підвищенні температури К (Н2О) зростає.
В чистій воді активності а(Н+) = а(ОН-) = 1*10-7 моль/л:
а(Н+) = а(ОН-) =
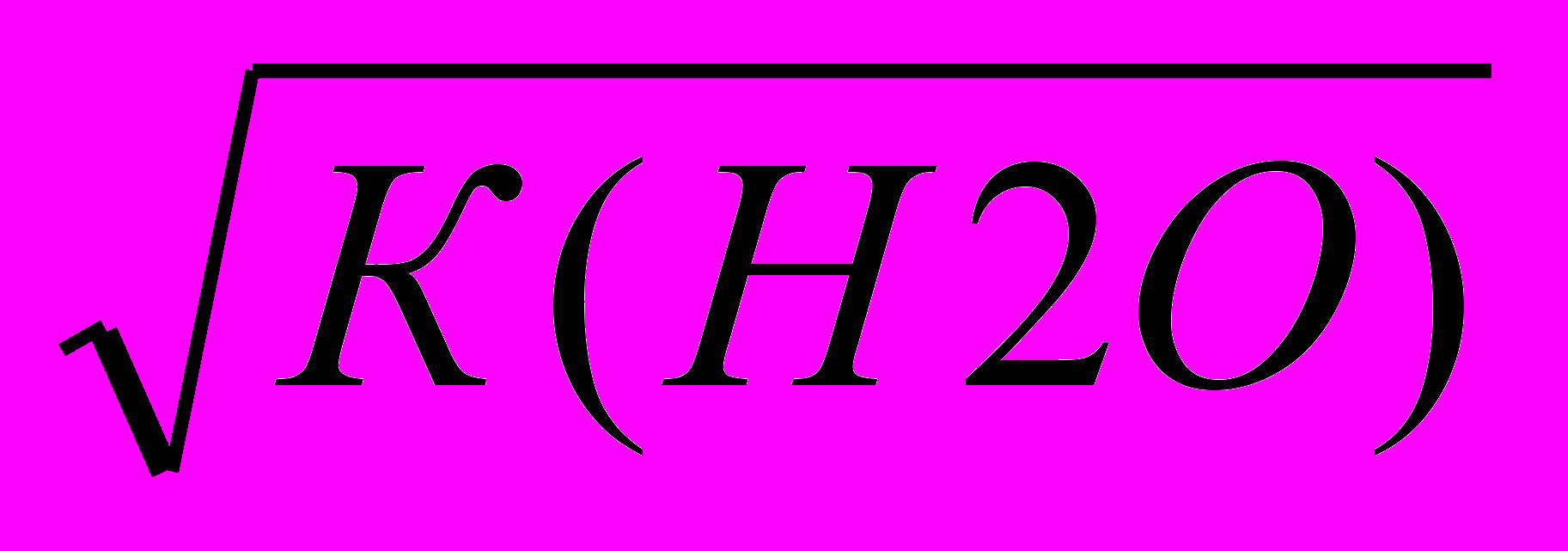 = √10-14 = 1*10-7
= √10-14 = 1*10-7 Якщо до чистої води додати стільки лугу, щоб концентрація гідроксид-іонів підвищилася, наприклад. до 10-4 моль/л, то концентрація [Н+] знизиться до 10-10 моль/л. Так що за законом діючих мас іонний добуток води залишиться рівним 1*10-14 моль2/л2.
Навпаки, якщо до чистої води додати стільки кислоти. Щоб концентрація іонів гідрогену підвищилася, наприклад, до 10-3 моль/л, то концентрація гідроксид-іонів знизиться до 10-11 моль/л, а іонний добуток води знову 1*10-14 моль2/л2.
Отже, знаючи концентрацію одного з цих іонів, завжди можна розрахувати концентрацію іншого іона. Як правило, для характеристики кислотності середовища використовують від’ємний десятковий логарифм активності (концентрації) гідроген-іонів, який називається водневим покажчиком рН середовища:
рН = -lg а(Н+)
або наближено рН = -lg а[Н+]
Нейтральне середовище рН = 7, кисле - рН < 7, лужне – рН >7.
Реакцію середовища можна характеризувати й гідроксид ним покажчиком : рОН = -lg а(ОН-)
або наближено рОН = -lg а[ОН-]
Тоді логарифмування виразу іонного добутку води дасть рН + рОН = 14.
В розчинах слабких кислот НА кислотно-основна рівновага ІІІ типу має вигляд
НА + Н2О ↔ Н3О+ + А- або НА ↔ Н+ + А-
Константа кислотної дисоціації Ка дорівнює:
Ка = [Н+][А-] / [НА].
У стані рівноваги [Н+]=[А-], тоді [НА] = (с(НА) - [Н+]). Підставивши це у вираз для константи, одержимо Ка = [Н+]2 / (с(НА) - [Н+]). Якщо загальна концентрація кислоти с(НА) більша як 0,01, значенням [Н+] у знаменнику можна зневажити і рівняння набуде вигляду Ка = [Н+]2 / с(НА).
Тоді [Н+] = √ Ка с(НА),
а рН = ½ р Ка – ½ lg с(НА), де р Ка = - lg Ка.
Для розчинів слабих основ формули для розрахунку рН вивести самостійно!
За аналогією до Ка – константою кислоти існує й Кb – константа основи. Між Ка та Кb є певний зв’язок: Ка Кb = 10-14
рКа + рКb = 14.
Обмінні реакції в розчинах.
При взаємодії різних електролітів відбуваються реакції обміну іонами:
А+В- + С+ D- ↔ С+В- + А+ D-
Відмінна риса обмінних реакцій – елементи, що входять до складу реагуючих речовин, не міняють свого ступеня окислення.
До обмінних реакцій у розчинах відносяться реакції: нейтралізації, гідролізу, осадження, розчинення, комплексоутворення.
Реакції між ними можна записувати в молекулярному, іонно-молекулярному та скорочено-іонному вигляді.