
М. Д. Благодатский, С. Н. Ларионов, А. А. Суфианов, Ю. А. Александров, М. А. Валиулин мальформация арнольда киари и сирингомиелия
| Вид материала | Документы |
- Идз №5 Индивидуальные задания из задачника Тюрин Ю. И., Ларионов В. В., Чернов, 268.29kb.
- Александров Д. Н. Личность и синдром предпринимателя, 72.06kb.
- Александров А. А, 49.6kb.
- Литература о Ломоносове М. В. в фондах библиотеки БашГУ, 55.77kb.
- Список книг, поступивших в библиотеку за июнь 2010, 960.34kb.
- В. А. Андреева // Кл рук. Науч метод журн для зам дир., кл рук., учителей нач шк., 163.33kb.
- «физиотерапия позвоночника», 197.9kb.
- Заслуженные люди визитная карточка деревни Александров Андрей Александрович «Заслуженный, 112.36kb.
- «не белы то снеги» в обработке А. В. Александрова александр Васильевич Александров, 14.7kb.
- Гостинично-ресторанный комплекс «Александров» приглашает к сотрудничеству. Всвязи, 660.43kb.
М.Д. БЛАГОДАТСКИЙ, С.Н. ЛАРИОНОВ, А.А. СУФИАНОВ, Ю.А. АЛЕКСАНДРОВ, М.А.ВАЛИУЛИН
МАЛЬФОРМАЦИЯ АРНОЛЬДА - КИАРИ И СИРИНГОМИЕЛИЯ
(сборник методических рекомендаций)
ИРКУТСК – 1995
Глава I.
МАЛЬФОРМАЦИЯ АРНОЛЬДА-КИАРИ.
ВВЕДЕНИЕ.
Цель настоящих рекомендаций - обратить внимание практических врачей на основные данные клиники, диагностики и принципы хирургического лечения патологии заднего мозга, вошедшей в литературу под названием "Мальформация Арнольда-Киари".
Необходимость освещения этих вопросов диктуется потребностями врачебной практики, так как, несмотря на столетнее изучение патологии заднего мозга, имеется явное несоответствие известного и изученного с представленными в отечественной литературе сведениями. В доступной практическому врачу литературе имеются лишь скудные данные, касающиеся отдельных вопросов изучения мальформации Арнольда-Киари (МАК), что явно недостаточно для полного представления о данной сложной патологии центральной нервной системы.
Впервые полное и детальное описание комплекса аномалий остеоневрального развития было сделано пражским профессором патологии Киари в 1891 и 1896 гг., который смог представить обсуждаемую патологию во всем спектре ее изменений.
Хронические грыжи мозжечка и ствола головного мозга в большое затылочное отверстие с клинически значимой неврологической симптоматикой наблюдаются как в младенческом, так и в зрелом возрасте. Дебют заболевания во многом определяется характером патологии заднего мозга, грыжи которого нередко сочетаются со стенозом сильвиева водопровода и выходных отверстий IV желудочка, незаращением центрального канала спинного мозга и другими пороками эмбриогенеза нервной системы, костными аномалиями образований краниовертебрального перехода, пороками и уродствами развития других органов и систем.
Клиническая значимость патологии остеоневрального развития этой области высока, однако неврологическая диагностика непроста. Клинический опыт показывает, что неврологические проявления МАК встречаются гораздо чаще, чем диагностируются. Это и побудило авторов изложить в свете современных данных основы патоморфологии, клинических проявлений, диагностики и лечения МАК.
В основу настоящих рекомендаций положен десятилетний опыт лечения 86 больных, 73 взрослых и 13 детей с МАК, а также анализ современной литературы.
ПАТОМОРФОЛОГИЧЕСКИЕ ИЗМЕНЕНИЯ В ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЕ ПРИ МАЛЬФОРМАЦИИ АРНОЛЬДА - КИАРИ
В настоящее время выделяют 3 основных типа мальформации. 1 тип МАК , по Киари, характеризуется смещением миндалин мозжечка через большое затылочное отверстие в заднее спинальное субарахноидальное пространство. Обычно миндалины достигают только уровня атланта или аксиса и такая патология носит название "первичная эктопия миндалин" (рис.16)
В редких случаях смещение миндалин может достигать дуг С5 или даже С6 позвонков. Наряду с каудальным смещением миндалин мозжечка при 1 типе МАК отмечается небольшое каудальное смещение продолговатого мозга. Отверстие Мажанди располагается ниже обычного уровня. Корешки каудальной группы черепно-мозговых нервов и трех верхних шейных сегментов спинного мозга имеют восходящее направление. Основу патологии этого типа МАК составляет эктопия миндалин мозжечка, которые частично или полностью тампонируют большую затылочную цистерну, как краниальный, так и спинальный ее отделы, и создают серьезные препятствия циркуляции ликвора из желудочковой системы в спинальное субарахноидальное пространство, Эктопия миндалин мозжечка в 10-15% случаев сочетается с облитерацией отверстия Мажанди, а в 20 - 25% с закрытием его частично перфорированной мембраной, что существенно отягощает патологию каудального смещения заднего мозга. В спинном мозге при первичной эктопии миндалин часто отмечается расширение центрального канала - гидромиелия, с реактивным глиозом или без него, Разрушение эпендимной выстилки интрамедуллярной полости приводит к развитию сирингомиелии. Длительное существование мозжечковой грыжи сопровождается реактивными изменениями мягкой мозговой и паутинной оболочек на уровне краниовертебрального перехода. Рубцовые процессы в оболочках создают дополнительные препятствия циркуляции спинномозговой жидкости и нередко являются отягчающим фактором патологии, приводящим к манифестации симптомов заболевания,
II тип МАК характеризуется более грубой и распространенной патологией нервной системы и наблюдается преимущественно в детском возрасте, у больных с врожденной спинномозговой грыжей в грудном, поясничном или крестцовом отделах позвоночника, Каудальное смещение продолговатого мозга при 11 типе МАК значительно выражено и сопровождается петлеобразованием. Продолговатый мозг прогибается назад и вниз, формируя цервикомедуллярный перегиб - "шпору", расположенную сзади и ниже верхних шейных сегментов спинного мозга." Эта "шпора" локализуется на уровне С2-С4, но может обнаруживаться и ниже (рис.2}
Степень смещения продолговатого мозга может быть различной. В большинстве случаев продолговатый мозг располагается ниже уровня большого затылочного отверстия, соответственно проекции дуг С1-С2 позвонков. IV желудочек удлинен до удвоенной длины, отверстия Мажанди и Люшка открываются в позвоночный канал, у 40% пациентов отверстие Мажанди облитерировано. Спинкой мозг при этом типе МАК также смещен
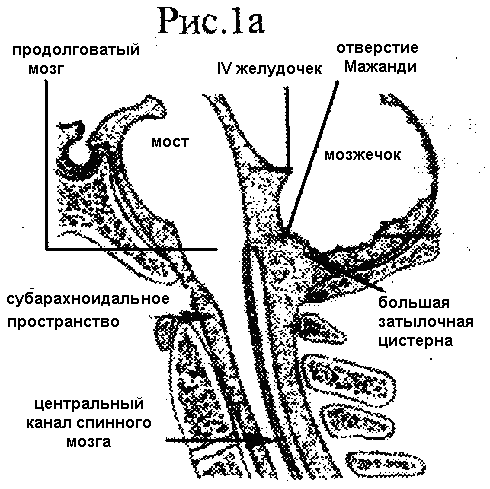
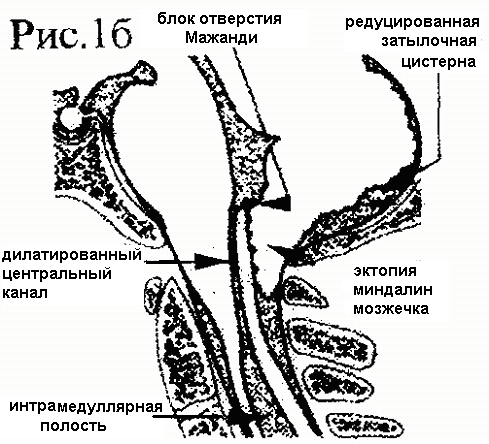
Рис.1. Анатомические взаимоотношения на уровне краниовертебрального перехода: а) норма; б) первичная эктопия миндалин мозжечка в заднее спинальное субарахноидальное пространство (по Williams, 1987г),
каудально и верхнешейные корешки имеют восходящее направление, соответствующие им сегменты спинного мозга становятся значительно короче, особенно СЗ-С6. укорочение высоты сегментов сочетается с уменьшением массы и количества нейронов. В 40-95% случаев наблюдается гидромиелия, а в 4-22% - цервикальная сирингомиелия. Полости спинного мозга выявляются как в грудном, так и в поясничном отделах, а в отдельных случаях полость может быть обнаружена в терминальной нити, которая, как правило, фиксирована и деформирована.
Мозжечок при II типе МАК гипоплазирован, нижний червь и гемисферы его плохо дифференцируются друг от друга, В отличие от МАК, тип I, при втором типе в позвоночный канал смещаются не только миндалины мозжечка, а и заостренный пальцеподобный вырост нижнего червя мозжечка, традиционно описываемый как "язык", “хвост","штифт". Длина его варьирует от 10 до 70 мм и более, располагается он на задней поверхности спинного мозга. Тип II МАК в большинстве случаев сопровождается выраженной гидроцефалией боковых желудочков мозга, и как следствие гидроцефалии происходит смещение большого мозга в среднюю черепную яму через расширенное отверстие мозжечкового намета. Изменения большого мозга не ограничиваются гидроцефалией и смещением, наблюдается распространенная складчатость, или микрогирия, гетеротопия кортикального серого вещества в стенки боковых желудочков мозга, утолщение межталамической спайки, гипоплазия серповидного отростка и мозжечкового намета, а также стеноз сильвиева водопровода. Для этого типа МАК характерна маленькая задняя черепная яма с низким расположением мозжечкового намета.
III тип МАК представляет собой нижнее смещение продолговатого мозга, сочетающееся с грыжей мозжечка и IV желудочка в шейно-затылочное менингоэнцефалоцеле или, другими словами, с шейной спинномозговой грыжей на уровне верхних шейных позвонков. Это крайне редкая и грубая форма врожденной патологии заднего, продолговатого и спинного мозга, при которой нарушаются взаимоотношения всех нервных структур задней черепной ямы и шейного отдела позвоночника. При III типе МАК обязательно присутствует
гидроцефалия и другие пороки развития большого мозга (рис.3).
ЭТИОЛОГИЯ И ЭПИДЕМИОЛОГИЯ МАЛЬФОРМАЦИИ АРНОЛЬДА-КИАРИ.
Для объяснения происхождения МАК предложены различные теории, которые рассматривают как врожденные, так и приобретенные эмбриологии Weed, считает, что эктопия заднего мозга является следствием нарушения перфорации крыши IV желудочка. В результате атрезии или стеноза отверстия Мажанди происходит накопление спинномозговой жидкости в полости IV желудочка, который в результате артериальной пульсации, передающейся с хориоидальных сплетений, действует как гидродинамический удар и смещает структуры заднего мозга каудально через большое затылочное отверстие, а также приводит к перерастяжению центрального канала спинного мозга.
Другие теории усматривают инициальный механизм патогенеза в фиксации спинного мозга, первичной дисгенезии ствола мозга с нарушением перфорации крыши IV желудочка и аномалиях развития понтомедуллярного перегиба и невральной трубки. Однако ни одна из предложенных теорий не может полностью объяснить происхождение всех типов МАК.
Эпидемиология МАК изучена недостаточно, но учитывая, что в 70- 90% МАК, тип I сочетается с сирингомиелией, а у 80-90% больных сирингомиелией выявляется МАК, то, вероятно, частота МАК, тип I, составляет 3,3-8,2 на 10000 населения, с известной вариабельностью по географическим зонам. Кроме того, II тип МАК имеет место у большинства новорожденных со спинномозговой грыжей и врожденной гидроцефалией, что составляет 1-2 случая на 1000 новорожденных. Соотношение мужчин и женщин приблизительно равное. Данных по изучению генетических аспектов мальформации практически нет. Известно только, что риск выявления МАК в семье со spina bifida в 3 раза выше, чем в общей популяции, а также отмечена большая частота выявления антигена локуса гистосовместимости НLА А9 у больных с сирингомиелией и костными аномалиями краниовертебрального сочленения.
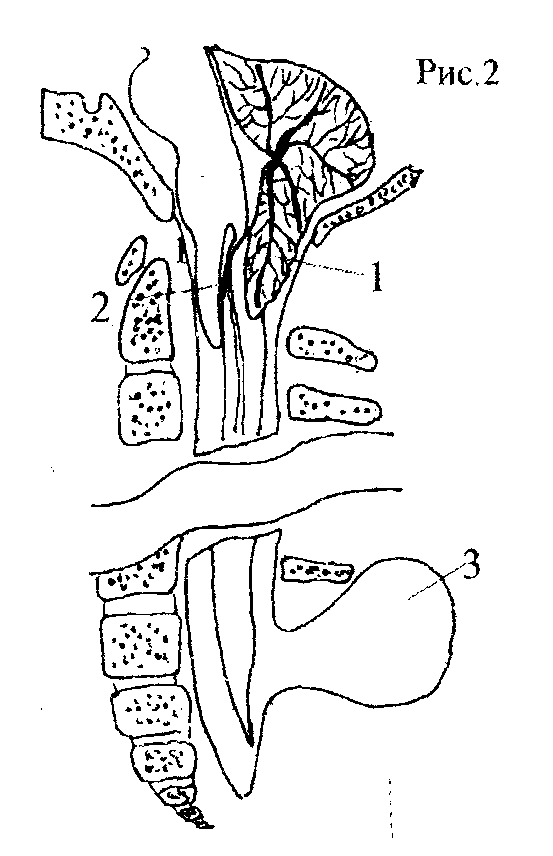
Рис 2.Анатомические взаимоотношения при МАК тип II.
1 - мозжечковая грыжа,
2 - цервикомедуллярный перегиб -"шпора",
3 - спинномозговая грыжа в пояснично-крестцовом отделе.
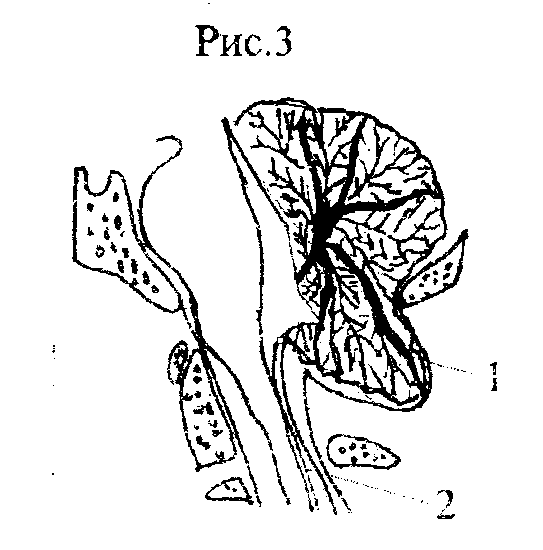
Рис 3. Анатомические взаимоотношения на уровне краниовертебрального перехода при МАК, тип III. 1-грыжа заднего мозга в шейно-затылочное менингоэнцефалоцеле, 2-расширенный центральный канал спинного мозга.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ МАЛЬФОРМАЦИИ АРНОЛЬДА-КИАРИ.
Клинические проявления МАК тип I преимущественно отмечаются у лиц молодого и зрелого возраста. Начало заболевания постепенное, реже острое. Когда мы говорим о начале заболевания в зрелом возрасте, то этот тезис, на наш взгляд, отражает не столько начало заболевания, сколько время установления диагноза. Между тем клинический опыт показывает, что при наличии грубой неврологической симптоматики диагноз МАК может быть установлен и в детском возрасте, Прежде всего это касается младенцев с врожденной гидроцефалией, у которых обструкция ликворных путей на уровне большой затылочной цистерны обусловлена МАК. Кроме того, у детей с врожденной патологией краниальных нервов (лицевого, отводящего, слухового, добавочного, подъязычного), проявляющейся гемиатрофиями лицевой мускулатуры, языка, кивательной мышцы, а также у детей с крупной головой, короткой шеей, врожденным сколиозом при целенаправленном обследовании может быть рано диагностирована МАК. Наконец, МАК в детском возрасте может себя проявить клиникой гипертензионного синдрома, такие больные иногда проходят в нейрохирургическом стационаре с диагнозом "арахноидит задней черепной ямы"
При отсутствии грубой неврологической симптоматики у отдельных сольных манифестация заболевания может быть в возрасте 15-25 лет. Первыми клиническими проявлениями у больных выступают шейно-затылочные боли, которые, как правило, интерпретируются как симптомы "шейного остеохондроза". Однако появление этих болей в молодом возрасте, связь болей с движением головы, усиление в момент кашля ("кашлевая боль"), чихания, натуживания в сочетании с расстройствами чувствительности в дерматомах С1- СЗ в виде гипо- или; гиперестезии, иногда с диссоциированным характером расстройств и продолжительность болевого синдрома более года склоняет диагноз в пользу эктопии миндалин мозжечка в большое затылочное отверстие.
В начале заболевания у таких больных возможны ремиссии, однако они непродолжительны и рецидив обычно сопровождается углублением неврологической симптоматики: зона болей распространяется на плечевой пояс, на этом же уровне определяются дополнительные участки гипестезии, появляются симптомы плече-лопаточного периартрита и вегетативно-трофические расстройства на кистях. Вовлечение в процесс зоны плечевого пояса усложняет проведение дифференциального диагноза с синдромами шейного остеохондроза (плечелопаточный болевой синдром, синдром Стейнброккера, звездчатого узла, заднешейный симпатический синдром). Однако и в такой клинической ситуации учет возраста пациента, непрерывной длительности болевого синдрома, выявления дизгенетических признаков (сколиоз, короткая шея, асимметрия черепа и др.), а также костных аномалий краниовертебрального перехода (платибазия, базилярная импрессия, ассимиляция атланта, аплазия дуги атланта и др.) дают достаточно оснований для клинического диагноза МАК. Контрастирование подоболочечных пространств или КТ подтверждают диагноз.
Большинство же случаев заболевания все-таки проявляются в зрелом возрасте. Клинические проявления полиморфны, выраженность симптомов колеблется от едва уловимых до грубого поражения продолговатого и спинного мозга, мозжечка и больших полушарий головного мозга. При всем многообразии причудливого переплетения симптомов, при МАК могут быть выделены следующие синдромы: спинальный пирамидный синдром, синдром центрального канала спинного мозга, бульбарный, мозжечковый и гипертензионно-гидроцефальный.
Спектр жалоб при МАК довольно широк и включает помимо отмеченных выше шейно-затылочных болей, жалобы на парестезии и гиперпатию в верхнем квадранте тела, двоение, головокружение, дизартрию, дисфонию, дисфагию, синкопальные состояния и пароксизмы ночных дыхательных расстройств.
При МАК тип I часто наблюдается спинальный пирамидный синдром, который выявляется у больных в различных вариантах: тетрапарез, парапарез, гемипарез, при двустороннем поражении характерна асимметрия паретических расстройств.
Наиболее грубое поражение спинного мозга в форме тетрапареза наблюдается при сочетании МАК и базилярной импрессии. Вентральное сдавление мозга зубом С2 может дать картину тяжелого поперечного поражения с появлением кистевых патологических рефлексов. Обычно пирамидные парезы конечностей сочетаются с симптомами страдания ствола и мозжечка. Преимущественное сдавление одной половины ствола дает клиническую картину ипсилатерального поражения краниальных нервов с ипси- или контралатеральным поражением конечностей.
При развившейся клинической картине МАК у большинства больных выявляются симптомы интрамедуллярного поражения, что свидетельствует о гидромиелии (двухсторонние диссоциированные зоны расстройств чувствительности с мягко выраженными пирамидными знаками), либо сирингомиелии, при которой уже выявляются симптомы переднерогового поражения спинного мозга.
Для бульбарного синдрома при МАК характерно одно- или двухстороннее страдание каудальной группы краниальных нервов (дисфония, дисфагия, атрофия мышц языка и т.д.). Однако в процесс могут быть вовлечены и другие краниальные нервы с V по VIII, что проявляется тригеминальной невралгией, парезами глазных мышц, головокружением. Характерен вертикальный нистагм при взгляде вниз.
Мозжечковый синдром проявляется чаще симптомами страдания флокулонодулярного комплекса (нарушение статики, походки, дизартрия). Могут наблюдаться и односторонние мозжечковые симптомы: дисметрия, диссинергия, интенционный тремор.
Церебральный гидроцефально-гипертензионный синдром при МАК имеет некоторые особенности, позволяющие его дифференцировать от внутричерепной гипертензии при опухолях головного мозга. Однако нужно отметить, что дифференциальный диагноз этих состояний не прост. В пользу происхождения этого синдрома от МАК свидетельствует относительная доброкачественность течения, компенсированность, длительность, слабо выраженный застой на глазном дне, имеющий тенденцию к обратному развитию под влиянием дегидратации, а также отсутствие в ликворе гиперальбуминоза, обычного спутника опухолевых заболеваний головного мозга, Существенное значение в дифференцировании этих состояний следует придавать симптомам интрамедуллярного поражения. И, наконец, правильной оценке шейно-затылочных болей, "кашлевой" головной боли и расстройствам чувствительности в трех первых шейных дерматомах. На первый взгляд эти симптомы имеют малое диагностическое значение, так как равным образом могут наблюдаться и при эктопии миндалин, и при их дислокации в случаях объемных процессов в задней черепной яме. Дифференциально-диагностическое значение имеет время появления шейно-затылочных болей и расстройств чувствительности в дерматомах С1-СЗ. При МАК это ранние, начальные симптомы заболевания, при объемных процессах это поздние симптомы, указывающие на дислокацию и, как правило, сочетающиеся с менингеальными симптомами.
Клинические проявления МАК тип II у детей со спинномозговой грыжей в люмбо-сакральном или грудном отделе позвоночника в первые недели жизни новорожденного могут быть выражены минимально, либо вообще не выявляются при обычном неврологическом исследовании. Наличие спинномозговой грыжи направляет в первые дни жизни лечебную тактику на оперативное вмешательство по закрытию менингомиелоцеле, которое, как правило, проводится без предварительной оценки состояния желудочковой системы и выявления возможной грыжи заднего мозга. К этому предрасполагает череп нормальных размеров в первые недели после рождения.
Однако вскоре после неонатального периода наблюдение за ребенком показывает, что он отстает в развитии, становится менее подвижным, испытывает трудности при кормлении, удержании головки и задержку темпа психомоторного развития. Голова ребенка начинает расти значительно быстрее, роднички расширяются, в них выбухает напряженный головной мозг - налицо симптомы прогрессирующей гидроцефалии. Неврологическая симптоматика в этот период отражает страдание нейрональных структур перерастянутых стенок боковых и III желудочка вследствие избыточного скопления ликвора. Мозжечковые симптомы у детей этого возраста выявить трудно. В то же время нередко выявляется страдание краниальных нервов, и как клиническое проявление поражения их – развитие ларингеального стридора вследствие двустороннего паралича абдукторов голосовых связей. Кроме того, страдание каудальной группы черепно-мозговых нервов может проявиться слабостью и атрофией мышц языка, дисфагией, слабостью лицевой мускулатуры, парезом отводящего нерва, снижением слуха.
Диагностика II типа МАК в первые месяцы жизни не представляет больших трудностей, если используется современная методика ультразвуковой секторальной эхоэнцефалографии через большой родничок, позволяющая визуализировать эту патологию без каких-либо дополнительных вмешательств для больного.
Диагноз же МАК I типа не прост. Проявления дизрафическрго статуса – первое, на что нужно обратить внимание. Со времени описания Бремером дизрафического состояния эта патология, как правило, отмечается при врожденных аномалиях центральной нервной системы. МАК не является исключением. Это и понятно, если принять во внимание, что МАК по своей природе есть также проявление дизрафии эмбриональной нервной трубки.
К внешним признакам дизрафии, часто наблюдающимся при МАК, относятся: кифосколиоз, воронкообразная грудь, асимметричный череп, короткая шея, низкая граница оволосения на шее, деформация стоп, аномалии сосков грудных желез и др. Важно подчеркнуть, что значение костных деформаций, выявляемых сразу же после рождения, для диагноза МАК велико.
Неврологическая диагностика МАК основывается на выявлении начальных проявлений: шейно-затылочные боли, "кашлевая"боль, синкопальные пароксизмы, расстройства чувствительности в трех верхних шейных дерматомах и в зонах Зельдера на лице. При развернувшейся неврологической картине для МАК характерно сочетание симптомов стволового, мозжечкового, гипертензионно-гидроцефального и пирамидного синдромов. Появление на этом фоне симптомов интрамедуллярного поражения с большей долей вероятности позволяет диагностировать МАК клинически.
Современная диагностика МАК немыслима без рентгенологического исследования, которое включает, помимо обзорной рентгенографии черепа и шейного отдела позвоночника, томографию краниовертебрального перехода, исследование ликвороносных пространств головного и спинного мозга рентгенопозитивными или рентгенонегативными контрастными средствами, а также КТ с метризамидным усилением и ЯМР. Проведенное рентгенологическое исследование должно дать объективную оценку следующих состояний:
1. Костные аномалии основания черепа и шейного отдела позвоночника (платибазия, базилярная импрессия, расширение позвоночного канала на уровне CI-CIII, конкресценция атланта, гипоплазия или аплазия дуги атланта, конкресценция шейных позвонков - синдром Клиппель-Фейля, кифосколиоз).
2. Проходимость выходных отверстий IV желудочка с вентрикулярной гидроцефалией.
3. Наличие интрамедуллярной полости.
4. Тип мальформации заднего мозга
Костные аномалии основания черепа и шейного отдела позвоночника наблюдается в 20-25% случаев МАК. Определение взаимоотношений костных структур на уровне краниовертебрального перехода производится на боковых рентгенограммах по линиям Чемберлена, Мак-Грегора, Мак-Рея и на фасных краниограммах по линиям Фицжгольда и де ля Пети.
Контрастное исследование ликворных пространств краниовертебрального перехода визуализирует эктопию миндалин мозжечка в заднее шейное спинальное субарахноидальное пространство, позволяет выявить симптом «флюктуации спинного мозга» при наличии интрамедуллярной полости, а также нарушение проходимости отверстия Мажанди и степень вентрикулярной гидроцефалии.
Существенное повышение уровня диагностики МАК произошло после внедрения в диагностический процесс КТ. Этот метод в сочетании с субарахноидальным введением метризамида позволяет визуализировать взаимоотношение нервных и костных структур, интрамедуллярные полости, установить тип МАК.
ЗАКЛЮЧЕНИЕ
Эффективность хирургической коррекции патологии заднего мозга, современные методы прямой визуализации структурных и функциональных изменений различных отделов ЦНС заставляют по новому взглянуть на проблему аномалий остеоневрального развития, в том числе и МАК. Вопросы клиники, диагностики и лечения МАК все чаще становятся предметом обсуждения в мировой неврологической и нейрохирургической литературе. Однако эти данные в большей своей мере труднодоступны практическому врачу.
Данные методические рекомендации восполняют этот пробел. Большой личный опыт авторов, с интраоперационной верификацией, а также патоморфологическим обоснованием выявляемых симптомов и синдромов позволяют рекомендовать конкретные критерии клинико-неврологической диагностики и современных инструментальных методов исследования. Использование их в практической работе позволят врачу своевременно диагностировать и лечить больных с МАК.