
 Авторефераты по темам >>
Разные специальности - [часть 1] [часть 2]
Авторефераты по темам >>
Разные специальности - [часть 1] [часть 2]
Математическая модель адсорбции простых молекул на наноструктурированных поверхностях и алгоритм поиска активных центров
Автореферат кандидатской диссертации

СТИШЕНКО Павел Викторович
Математическая модель адсорбции простых
молекул на наноструктурированных поверхностях
и алгоритм поиска активных центров
Специальность 05.13.18 - Математическое моделирование, численные методы
и комплексы программ
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата физико-математических наук
Омск-2011
Работа выполнена в Государственном образовательном учреждении
высшего профессионального образования
Омский государственный технический университет
Научный руководитель:
доктор химических наук МЫШЛЯВЦЕВ Александр Владимирович
Официальные оппоненты:аа доктор физико-математических наук,
профессор БЫКОВ Валерий Иванович
Ведущая организация:
кандидат физико-математических наук, доцент ПИЧУГИН Борис Юрьевич
Институт вычислительной математики и математической геофизики СО РАН, г. Новосибирск
Защита состоится 16 июня 2011 г. в 16:00 на заседании диссертационного совета ДМ 212.179.07 при Омском государственном университете им. Ф.М. Достоевского по адресу 644099, г. Омск, ул. Певцова, 13.
С диссертацией можно ознакомиться в библиотеке Омского государственного университета им. Ф.М. Достоевского.
Автореферат разослан
мая 2011 г.
|

у(А4<о |
Ученый секретарь
диссертационного совета,
кандидат физико-математических наук
& A.M.
Семенов
Актуальность работы. Современные технологии создания и исследования поверхностных наноструктур открыли новые возможности для управления свойствами гетерогенных катализаторов. В 1987 году Харута показал высокую каталитическую активность металлических наночастиц (М. Haruta et al // Chem. Lett. Ч 1987. Ч Vol. 16, no. 2. Ч Pp. 405^-08.). Это стимулировало активные исследования в этой области, и в настоящее время подавляющее большинство промышленных и автомобильных катализаторов представляют собой наноча-стицы переходных металлов, нанесенные на поверхность пористой подложки. Выявлен целый ряд явлений и эффектов, которые могут объяснять повышенную реакционную способность наночастиц. Например: наличие большого числа низкокоординированных (low-coordinated) атомов на ребрах, возле ступенек и дефектов решетки, на высокоиндексных гранях; диффузия адсорбированных молекул между гранями и подложкой; наличие границы металл-подложка, на которой могут создаваться благоприятные для реакции условия. Исчерпывающее описание таких эффектов можно найти, например, в обзоре В. R. Cuenya // ThinSolidFilms. Ч 2010. Ч Vol. 518, no. 12. Ч Pp. 3127-3150. Из этого обзора следует, что, несмотря на широкое использование катализаторов на основе наночастиц, степень влияния этих эффектов на конечную скорость реакции остаётся невыясненной и разработка таких катализаторов по-прежнему ведётся методом проб и ошибок.
Для того чтобы описать процессы, происходящие на наноструктурирован-ных поверхностях, одних экспериментов недостаточно. Необходимо разработать математические модели, которые бы позволили обобщить и интерпретировать экспериментальные данные и понять природу изучаемых явлений.
Цель диссертационной работы состоит в разработке математической модели, описывающей адсорбцию молекул на поверхности нанесенных металлических наночастиц, и алгоритмов поиска их равновесной формы и покрытия.
Для достижения поставленной цели необходимо решить следующие задачи:
- Разработать математическую модель адсорбции молекул на нанострукту-рированных поверхностях с учетом геометрии и энергетики множества типов центров адсорбции.
- Разработать алгоритм моделирования элементарных физико-химических процессов в адсорбционном слое на реалистичной поверхности наноча-стицы.
- Разработать эффективный алгоритм поиска равновесной формы и морфологии поверхности наночастицы с учётом разнообразия адсорбционных центров.
3
- Реализовать разработанные алгоритмы в виде компьютерной программы.
- Подтвердить предсказательную силу модели, воспроизведя в её рамках известные экспериментальные зависимости.
Научная новизна работы:
- Предложена математическая модель адсорбции на активные центры с различной геометрией и энергией на сложноструктурированной поверхности кристалла с произвольной решёткой.
- В рамках предложенной модели разработан алгоритм поиска центров адсорбции разных типов со сложностью О(М).
- Разработан алгоритм расчета изменения энергии с использованием неаддитивных потенциалов со сложностью О(М).
- На основе этих алгоритмов разработан программный продукт, позволяющий решать задачу о равновесном покрытии наночастиц адсорбентом.
- С помощью разработанного программного продукта воспроизведено изменение доли моноцентровых (atop) и мостиковых (bridge) центров адсорбции в общем покрытии наночастиц платины молекулами СО при изменении давления газа.
Теоретическая значимость
Разработана математическая модель адсорбции молекул на сложных нано-структурированных поверхностях с применением метода Монте-Карло, учитывающая геометрию и энергетику множества активных центров и алгоритм решения задачи о равновесном покрытии в рамках этой модели.
Практическая значимость
Полученные в ходе моделирования данные показали, что покрытие платиновых наночастиц адсорбированным СО в значительной степени определяется энергетикой и геометрией адсорбции на гранях частицы. Выполненные оптимизации алгоритмов позволили значительно ускорить нахождение равновесного состояния систем, что даёт возможность исследовать адсорбцию на сложной поверхности, сформированной из тысяч атомов.
Основные положения, выносимые на защиту
На защиту выносятся:
1) математическая модель адсорбции молекул на металлических наночастицах;
4
- алгоритм поиска центров адсорбции на сложных наноструктурированных поверхностях;
- алгоритмы расчета энергии с помощью неаддитивных потенциалов, работы со списками активных атомов и вакансий и способы распараллеливания процесса моделирования диффузии.
Методы исследования
Модель адсорбции разработана на основе методов статистической физики и теории случайных процессов. При разработке и реализации алгоритмов моделирования использовались методы теорий алгоритмов, клеточных автоматов и экспериментальной алгоритмики. Для оценки предсказательной силы модели результаты моделирования сравнивались с известными из литературы данными инфракрасной спектроскопии.
Апробация работы
Основные результаты диссертации докладывались на следующих конфе
ренциях и семинарах: IV Всероссийская научная молодежная конференция
Под знаком Z (Омск, 2007 г.); Tenth International Symposium on Heteroge
neous Catalysis: Quo Vadis, Catalysis?аа (Болгария, Варна, 2008 г.); XVIII
International Conference on Chemical Reactors CHEMREACTOR-18 (Мальта,
Аура, 2008 г.); 51-я научная конференция МФТИ, (Москва, 2008 г.); The Sev
enth International Symposium on Effects of Surface Heterogeneity in Adsorption
and Catalysis on Solids ISSHAC-7 (Польша, Казимеж-Дольный, 2009 г.); Меж
дународная научная конференция Параллельные вычислительные технологии
2010 (Уфа, 2010 г.); семинар в Омском филиале Института математики им.
С.Л. Соболева СО РАН (Омск, 2010 г.); семинар по клеточным автоматам в
Институте вычислительной математики и математической геофизики СО РАН
(Новосибирск, 2011 г.).
Публикации
Материалы диссертации опубликованы в 9 печатных и электронных изданиях, из них 1 статья в международном рецензируемом журнале Applied Surface Science, 1 статья в журнале Омский научный вестник (включен в список ВАК) и 6 публикаций в сборниках трудов конференций и тезисов докладов. Программа "Monte Carlo simulation tool for metallic nanoparticles" зарегистрирована в реестре программ для ЭВМ ФГУ ФИПС под номером 2009613884.
Структура и объем диссертации
Диссертация состоит из введения, трех глав, заключения и библиографии. Общий объем диссертации 166 страниц, включая 23 рисунка. Библиография включает 177 наименований.
5
Основное содержание работы
Во введении обоснована актуальность исследования, сформулированы цели и задачи работы, объяснен выбор модели, описано содержание глав диссертации и приведены основные результаты работы.
В первой главе описаны особенности современного катализа, приведен обзор основных методов молекулярного моделирования. Описаны основные математические модели адсорбции на гетерогенных поверхностях и алгоритмы вычислений в рамках этих моделей. Указано, что наилучшие результаты достигнуты в рамках псевдотрёхмерной клеточно-автоматной модели на основе кристалла Косселя с применением метода Монте-Карло и в рамках непрерывной модели с применением теории функционала плотности (ТФП). Также описаны наиболее часто используемые эмпирические потенциалы энергии, сделаны обзоры работ по моделированию наночастиц и способов моделирования адсорбции на неоднородных поверхностях, обоснована необходимость разработки модели адсорбции на металлических наночастицах. Далее описаны основы наиболее широко используемых методов молекулярного моделирования: ТФП, молекулярной динамики и Монте-Карло. Методы ТФП являются наиболее точными и используются для относительно небольших систем, например для расчёта энергии и геометрической конфигурации адсорбции отдельных молекул, но они слишком затратны для систем из тысяч атомов. Методы молекулярной динамики позволяют моделировать системы, состоящие из большого числа атомов, но на временных интервалах до несколько сотен наносекунд, тогда как уравновешивание систем с участием наночастиц может занимать несколько часов и даже дней. В параграфе 1.3.3 дан краткий обзор свойств метода Монте-Карло, применительно к задачам поиска равновесной формы и покрытия наночастиц. Подчеркнуто, что метод позволяет как находить равновесное состояние систем с большим временем релаксации, так и исследовать кинетику длительных химических процессов.
Параграф 1.4 посвящен эмпирическим потенциалам межатомного взаимодействия, используемым в молекулярном моделировании. Потенциалы могут быть парными (Леннарда-Джонса) и многочастичными (ЕАМ/МЕАМ, Саттона-Чена, ближних связей). Потенциалы первого типа дают меньшую точность, но сложность их вычисления равна 0(N2), где N - число атомов в системе. Для переходных металлов, из которых состоят наночастицы катализаторов, чаще используются более точные многочастичные потенциалы, сложность вычисления которых равна 0(N3). При моделировании методом Монте-Карло на расчет изменения энергии АЕ уходит большая часть процессорного времени, поэтому эффективная реализация этой процедуры имеет большое значение. Обычно не учитываются взаимодействия между атомами, расстояние между которыми
6
превышает некоторый заданный радиус отсечки. Тогда в качестве N выступает число атомов в пределах этого радиуса.
В параграфе 1.5 приведен обзор методов, использованных в последних работах по поиску равновесной формы наночастиц. Показано, что для наночастиц из сотен и тысяч атомов, представляющих интерес для катализа, чаще всего применяется метод Монте-Карло.
Из оценки свойств основных методов молекулярного моделирования и обзора работ по моделированию наночастиц и моделей адсорбции сделан вывод, что для решения поставленных задач наиболее подходит метод Монте-Карло. Более подробное описание метода приведено во второй главе в параграфе 2.1. Моделирование адсорбции методом Монте-Карло выполняется в рамках большого канонического ансамбля, то есть при переменном числе атомов в системе (NN) и при постоянных значениях химического потенциала (р.), объема (V) и давления (Г).
В конце первой главы делается вывод, что существующие модели не позволяют вычислять равновесное покрытие сложных поверхностей с учётом разнообразия активных центров адсорбции.
Вторая глава посвящена подробному описанию метода Монте-Карло, разработанной математической модели адсорбции и алгоритмам вычисления равновесной формы и покрытия наночастиц на её основе. Приведены доказательства корректности разработанного алгоритма моделирования адсорбции, константной сложности операций со списками активных ячеек, линейной сложности предложенных алгоритмов вычисления изменения энергии для неаддитивных потенциалов и поиска активных центров адсорбции.
В параграфе 2.1 приведены основные предположения модели и её входные данные. Модель строилась исходя из существующих на сегодняшний день теорий о каталитической активности наночастиц и экспериментальных данных об адсорбции газов на их поверхности.
Предположения модели:
- адсорбирующая поверхность может быть описана в рамках модели решёточного газа (МРГ);
- адсорбция может происходить только на множестве активных центров (АЦ), расположенных на поверхности адсорбента;
- каждый А - может быть однозначно отнесён к одному из небольшого количества типов;
- каждый тип А - однозначно определяется взаимным расположением адсорбированной молекулы и атомов адсорбента в пределах 1-3 координационных сфер;
7
5)а каждому типу А - соответствует определённое значение энергии адсорб
ции;
6)а типы А - инвариантны относительно вращений и параллельных переносов.
Входные данные модели:
- тип кристаллической решётки наночастицы;
- тип и параметры потенциала межатомного взаимодействия внутри частицы и между наночастицей и подложкой;
- список типов активных центров, задаваемых геометрией и энергией адсорбции каждого типа;
- радиус потенциала жёстких сфер для латеральных взаимодействий между адсорбированными молекулами.
В параграфе 2.2, следуя учебникам D. Frenkel, A. Leach, М. Newman, приведено детальное описание метода Монте-Карло и алгоритма Метрополиса как основы для разработанных нами алгоритмов. Описаны физические основания метода, детально описаны алгоритмы моделирования адсорбции в нерешёточных моделях по методу Нормана-Филинова, а также адсорбции и диффузии в рамках МРГ.
Применение метода Монте-Карло для решения задачи поиска равновесного состояния термодинамических систем основано на предположении о том, что любое из состояний системы с одинаковыми значениями числа молекул (N), объема (V) и потенциальной энергии (Е) является равновероятным. Из этого предположения можно вывести вероятность найти систему в любом из возможных состояний i:
=аа exp(-Ej/kBT) ZjexpiEj/ksT)'
Здесь jпробегает по всем возможным состояниям системы. Выражение, стоящее в знаменателе, называется большой статистической суммой. Из-за огромного числа состояний её прямое вычисление является очевидно безнадежной задачей.
Для получения приближения некой величины f(i), зависящей от состояния системы и усредненной по вероятности обнаружить систему в каждом из состояний, воспользуемся следующей формулой:
^ХЛОР=Itjf(Oexp(-Ei/kBT)
YuiPi%кхр(-Е/квТ)
8
Здесь iпробегает не по всем состояниям, а по случайному их подмножеству. Важно, что вместо абсолютных вероятностей /?; в этом выражении используется так называемый фактор Болъцмана ехр(-Е/квТ).
Приближение 2 тем ближе к точному решению, чем больше сумма YjiP(i)-Поэтому нужно так организовать выборку случайных состояний, чтобы в неё попали наиболее вероятные из них, то есть с наименьшими энергиями Et. В работе The Monte Carlo Method / N. Metropolis, S. Ulam // J. of Am. Stat. Assoc. Ч 1949. Ч Vol. 44, no. 247. Ч Pp. 335-341. Метрополис и Улам предложили использовать для этого следующий марковский процесс:
- В окрестности текущего состояния iслучайным образом выбирается некоторое состояние j, обладающее заведомо низкой энергией. Обозначим вероятность выбрать состояние jиз состояния iкак а,ц.
- С некоторой вероятностью pij, зависящей от изменения энергии системы АЕ = Ej- Е(, текущим становится состояние jили с вероятностью 1 - р^ состояние не меняется.
Этот алгоритм определяет матрицу переходов марковского процесса , где jiij= etij* Pij. В соответствии с теоремой о предельных вероятностях, многократное повторение шагов этого процесса приводит систему к равновесию с распределением рцт = /шгт_>ооР(1) * т. Тогда справедливо условие
Yjpi*nij = pj*YjnjiVj,аа (3)
называемое условием баланса. Чаще применяют более сильное условие детального баланса:
Р0(] = pjiijiVi, j.(4)
Из уравнения 1 следует, что
Pil-(Ei-Ej)\
Ч = ехр ----- Ч-Ча ,а (5)
Pi\а квТа J
или
^ = ехр(-(^-^У (6)
ацРц\а квТа )
Очевидно, что существует множество способов задать матрицы а и р. В оригинальной работе Метрополиса матрица а считалась симметричной, а матрица р определялась по формуле
Хаа L(-(Ej-EdW
Pij = min\l;exp\ЧЧЧII. (7)
9
Важно, что подмножество состояний, из которых делается выбор на каждом шаге, ничем не ограничено, и марковский процесс не обязан соответствовать реальной кинетике процесса уравновешения системы. Именно это позволяет быстро находить равновесные состояния систем с большим временем релаксации. Матрицы а и р зависят от используемого статистического ансамбля и модели исследуемой системы. В параграфе 2.2 описаны марковские процессы, применяемые при моделировании адсорбции в нерешёточной модели и диффузии атомов.
В параграфе 2.3 описаны разработанные нами способы повышения скорости поиска равновесной формы и морфологии поверхности наночастиц, а также приведены доказательства константной и линейной сложности предложенных алгоритмов:
- ускорение вычислений изменения энергии для непарных потенциалов за счет хранения аддитивных членов (параграф 2.3.1);
- специализированные контейнеры для списков активных ячеек (параграф
2.3.2).
Обычно при моделировании диффузии выбор атома и вакансии делается не из всей решетки, а из списков активных ячеек. В этих списках хранятся координаты атомов и вакансий, для которых вероятность перехода наиболее высока - атомы первых двух поверхностных слоев наночастицы и вакансии у её поверхности и на подложке. Очевидно, что необходимо постоянно поддерживать актуальность этих списков. Насколько нам известно, сегодня нет общепринятых структур данных, используемых для списков активных атомов и вакансий. В то же время выбор эффективной структуры данных для этих целей значительно влияет на скорость моделирования. При моделировании с ними выполняются следующие операции:
- выбор одного элемента в соответствии с равномерным распределением вероятностей;
- добавление и удаление элементов после каждого успешного шага.
Массив, дерево или список имеют сложность не меньше 0(logN) как минимум для одной из этих операций, но в решеточной модели возможно обеспечить константную сложность для всех. В нашей реализации алгоритма этот контейнер состоит из двух массивов R и L и индекса TAIL. Массив R имеет заведомо достаточную длину, чтобы вместить все активные атомы и содержит их координаты. Массив L является развёрткой кристаллической решётки, в которой находятся атомы наночастиц. Каждый элемент массива L содержит либо индекс элемента в массиве R, либо -1, означающее, что в данной ячейке решётки нет
10
активного атома. Массив R содержит индексы элементов массива L. При этом соблюдается условие, что R[i] = j, если и только если L[j] = i. В массиве R элементы, ссылающиеся на активные атомы решётки, расположены непрерывно от начала массива и до элемента, на который указывает TAIL. Остальные элементы массива не используются. Когда в ходе моделирования число активных атомов меняется, то TAIL сдвигается в ту или другую сторону.
В таком контейнере операции вставки, удаления и выбора случайного элемента могут имеют сложность 0(1). Действительно, зная координаты атома, можно определить его индекс iв развёртке решётки L. Чтобы вставить новый активный атом, потребуется
- присвоить L[i] текущее значение TAIL,
- присвоить R[TAIL] значение i,
- увеличить TAIL на 1.
Чтобы удалить имеющийся атом, нужно выполнить следующие операции:
- присвоить R[L[i]] значение R[TAIL],
- уменьшить TAIL на 1,
- присвоить L[i] значение -1.
Выбор случайного активного атома выполняется генерацией случайного числа от 1 до TAIL. Очевидно, что все эти процедуры действительно имеют сложность 0(1).
Расчет изменения энергии системы в результате акта диффузии - это
самый затратный с вычислительной точки зрения шаг алгоритма. Приведем уравнения энергии для потенциала ближних связей:
Nаа (,--------- \
Е'total = 2jаа \jRiJ~а S^jBiJаа ж№
Здесь N - общее количество атомов, Rtjи Btj- параметры отталкивания и притяжения соответственно между атомами iи j. Уравнение (8) можно переписать в следующем виде:
Etotal = YuYuR^-YuSL^(9)
11
или
Е
total
Ч Ej? Ч En.
(10)
Пусть AEtotai- изменение общей энергии системы при одном акте диффузии, AEr- изменение составляющей отталкивания, а АЕВ - изменение составляющей притяжения. Если в радиус учитываемого межатомного взаимодействия (радиус отсечки) попадает М атомов (значение М зависит от типа решетки и требуемой точности вычислений), то при одном акте диффузии в выражении для Erизменится не более чем 2М значений Rtj. Тогда при перемещении /-го атома для изменения энергии AERопределяется по следующей формуле:
2М
R |
АЕ
Rn).
(И)
Здесь Rnn- энергия отталкивания между атомами iи / после акта диффузии, a iпробегает всех соседей /-го атома в пределах радиуса взаимодействия до и после акта диффузии.
Значение АЕв не может быть вычислено таким же образом из-за наличия корня в выражении для Ев. Поэтому необходимо пересчитывать значения всех корней, под которыми изменилась хоть одна Btj
Аналогично, здесь Впи означает энергию отталкивания между атомами iи jпосле акта диффузии, a iпробегает всех соседей /-го атома в пределах радиуса отсечки до и после акта диффузии. В свою очередь jпробегает по всем соседям каждого /-го атома. Ключевым отличием от выражения для AERявляется то, что мы не можем посчитать изменение корня из суммы, зная лишь изменение одного элемента суммы. Поэтому мы вынуждены полностью пересчитывать энергию каждого соседа /-го атома, что добавляет ещё один вложенный цикл в алгоритм вычисления АЕВ. Таким образом, для непарных потенциалов сложность расчета изменения энергии равна 0(М2).
Для снижения этой сложности мы применили прием, называемый мемо-изацией. Если для каждого /-го атома хранить актуальное значение суммы 13, то при перемещении одного из атомов (пусть с индексом /) можно вычислить её
12
изменение как разницу Впй - Вц. Пусть
м
BiS=аа J]а ВФ(13)
тогда
АЕв= Z (^Bts-Вй+в - J**) ж(14)
Соответственно для всех соседей /-го атома потребуется пересчитать Zfo после успешного акта диффузии:
BniS=BiS-Bil+Bna.(15)
Очевидно, что, в отличие от полного пересчета суммы, эта операция имеет сложность не 0(М2), а О(М), то есть такую же, как и у парных потенциалов. Конечно, необходимо поддерживать актуальность суммы для всех атомов частицы, что требует дополнительных затрат. Их величина напрямую зависит от доли успешных шагов. Поэтому не следует ожидать ускорения вычислений в М раз.
Мы разработали два способа распараллеливания алгоритма диффузии и выполнили экспериментальную оценку их эффективности с учетом предложенных выше оптимизаций и структур данных. Были реализованы два возможных способа: основанный на пространственной декомпозиции области моделирования и основанный на параллельной оценке нескольких шагов диффузии.
В первом случае пространство моделирования случайным образом разбивалось на несколько независимых областей (по одной на поток моделирования). На границах каждой области выделялся слой ячеек решетки, находящихся в пределах радиуса отсечки от соседней области. Этот приграничный слой непосредственно не участвовал в моделировании диффузии, но для этих ячеек изменялись значения хранимых аддитивных составляющих энергии В($. Пример такого разбиения приведен на рисунке 1. Переразбиение поверхности наноча-стицы на независимые области происходило каждые 10 шагов Монте-Карло случайным образом.
Другой подход к распараллеливанию вычислений основан на особенности моделирования металлических наночастиц в рамках решеточной модели. Из-за того, что расположенные в узлах кристаллической решетки атомы имеют большую энергию связи, вероятность успешного их перемещения весьма низкая. Это позволяет оценивать сразу несколько пробных шагов. При этом существует небольшая вероятность события, когда успешными окажутся сразу
13
Рис. 1. Пример разбиения поверхности наночастицы на независимые области |
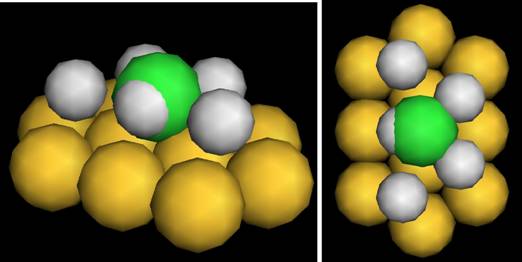
Рис. 2. Конфигурация адсорбции на грани (111). Тёмная (зелёная) сфера - адсорбированный атом, большие светлые (жёлтые) сферы - атомы частицы, маленькие светлые (белые) сферы обозначают требуемые вакансии в решётке
несколько шагов. В случае моделированной нами нанесенной частицы платины такие ситуации возникали не более чем для 5% шагов.
Далее во второй главе в параграфе 2.4 Моделирование адсорбции описаны разработанные нами структуры данных и алгоритмы, используемые для моделирования адсорбции на полученной поверхности наночастицы. Показано, что при использовании алгоритма Метрополиса для моделирования адсорбции в рамках МРГ, чтобы обеспечить симметричность матрицы априорных вероятностей от, требуется гарантировать равную вероятность выбора каждого А - для актов адсорбции,десорбции или диффузии. Это легко сделать для адсорбирующей поверхности с регулярной структурой или с одним типом АЦ. В случае же наличия множества типов А - и сложной структуры поверхности эта задача становится нетривиальной.
14
Имеющиеся данные об энергиях адсорбции привязаны к типам активных центров, которые задаются в терминах граней ((100), (111), (211) и т. д.) и типов адсорбции (моноцентровая - atop, мостиковая - bridge, трехкоординатная - in-hollow и т.д.). Для описания активных центров мы построили пространственные конфигурации, состоящие из внутренних координат атомов и вакансий наночастицы и молекулы адсорбата - внутренние конфигурации (рисунок 2).
Алгоритм поиска активного центра по внутренней конфигурации основан на рекурсивном обходе в глубину дерева соседних ячеек решётки с сопоставлением каждой ячейки с элементами внутренней конфигурации. Если на каком-то шаге для элемента конфигурации не удалось найти ячейку с подходящим содержимым и на требуемом расстоянии, то происходит возврат к предыдущему элементу конфигурации. Такой поиск с возвратом требует порядка 0(2N) операций. Как минимум, таким образом необходимо искать первые три ячейки. Также вычисление углов и декартовых координат требует использовать операции с плавающей точкой, что негативно влияет на производительность алгоритма.
Поэтому для каждой ячейки решетки был построен список соседних атомов. Этот список упорядочен таким образом, что для любой ячейки вектор до -то соседа будет одним и тем же для одинаковых i. Важно, что векторы от атома до его -то соседа одинаковы и по длине, и по направлению. Таким образом, списки соседей полностью описывают топологию решётки.
Поскольку А - определятся именно взаимным расположением атомов в небольшой окрестности, то можно использовать номера ячеек в списках соседей для задания пространственных конфигураций в решетке. Для их представления в памяти ЭВМ мы выбрали дерево, в узлах которого находились атомные маски, а ветви были отмечены целыми числами так, чтобы обозначать, каким по номеру соседом является дочерний узел по отношению к родительскому. Будем называть такие конфигурации решёточными. Благодаря такому представлению конфигурации активного центра его поиск на поверхности наночастицы сводится к обходу дерева решёточной конфигурации без возвратов, то есть требует проверки только содержимого 0(N) ячеек решётки. При этом не требуется работать с внутренними координатами, а следовательно, и с числами с плавающей точкой. При генерации решёточных конфигураций из внутренних, выполняется их попарное сравнение на эквивалентность относительно параллельных переносов. Таким образом, каждому А - решётки соответствует одна и только одна пара ячейка-решёточная конфигурация. Тогда, для того, чтобы обеспечить равновероятность выбора А - и симметричность матрицы а, достаточно выбирать в соответствии с равномерным распределением случайные ячейки и решёточные конфигурации.
15
Разработанная математическая модель реализована в виде программы на языке C++ с использованием стандартной библиотеки (списки, динамические массивы и ассоциативные контейнеры) и некоторых библиотек Boost (управление памятью, сериализация, настройки программы, генерация файлов для визуализации). В качестве генератора случайных чисел использовалась реализация вихря Мерсена из библиотеки Агнера Фога ( В качестве моделируемой системы была выбрана нанесенная наночастица платины из 4033 атомов (до 6 нм в диаметре для нанесённых частиц), адсорбирующая молекулы СО. Для описания взаимодействия атомов частицы был взят широко используемый потенциал ближних связей. С использованием разработанного алгоритма, была найдена равновесная форма и морфология поверхности наночастицы. Состояние равновесия определялось по колебаниям на графике энергии системы (рисунок 3). Для ускорения процесса уравновешивания применялась техника симуляции отжига (simulated annealing). Температура системы проходила три уровня: 1978 К, 1163 К и 581 К. На каждом уровне выполнялось достаточное число шагов, чтобы устранить влияние начальной формы частицы и убедиться в том, что не происходит значительных изменений уровня энергии. Экспериментально было установлено, что для этого достаточно 6000 шагов при температуре 1978 К и по 1000 при температуре 1163 К и 581 К.
Была измерена скорость алгоритма при двух стратегиях обновления списков активных атомов и вакансий: каждые 10 шагов Монте-Карло (каждый шаг включает в себя 720000 попыток акта диффузии) и после каждого успешного акта диффузии. Первая стратегия использовалась при пространственном распараллеливании алгоритма, а вторая - при одновременных попытках. При работе с одним потоком существенных отличий между этими стратегиями выявлено не было - небольшое преимущество в скорости первой стратегии компенсировалось меньшей устойчивостью графика энергии (рисунок 3).
Всего для нахождения равновесного состояния частицы с использованием разработанного алгоритма требовалось не более 10000 шагов Монте-Карло, или 10 часов процессорного времени, но в большинстве случаев равновесное состояние достигалось вдвое меньшим числом шагов. Такая производительность алгоритма приемлема для вычислительных экспериментов.
16
EE,eV------ 2000 T,K - _ lg00 -аа 1600 |
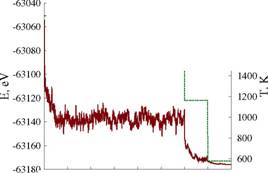
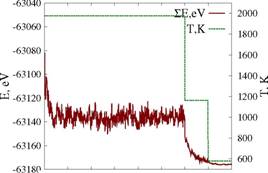 0 1000 2000 3000 4000 5000 6000 7000 8000
0 1000 2000 3000 4000 5000 6000 7000 8000
MCS
0 1000 2000 3000 4000 5000 6000 7000 8000
MCS
Рис. 3. Энергия и температура в ходе моделирования нанесенной НЧ платины размером в 4033 атома (слева - обновление каждые 10 шагов, справа - каждый успешный акт)
 700-
700-
>Х
ч
* 600 ж к
Пространственная декомпозиция Параллельные попытки
2а 4а 6
Число потоков
ж700 ж600 500 400 300 200 100 жО
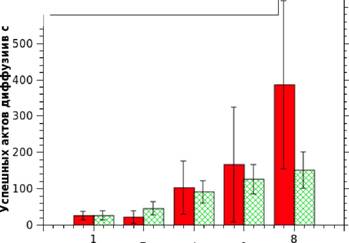
Рис. 4. Масштабируемость методов распараллеливания моделирования процесса диффузии при температуре 1160 К (число успешных шагов в секунду в зависимости от числа задействованных ядер).
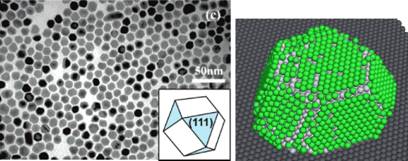
Рис. 5. Модельная частица платины, подложка, покрытая наночастицами платины, модельная частица платины (белые сферы), покрытая молекулами СО (зеленые сферы)
Чтобы оценить влияние предложенной оптимизации расчета энергии с помощью многочастичных потенциалов, мы реализовали алгоритм без этой оптимизации и измерили разницу в производительности. Результаты профилирования показали, что мемоизация аддитивных членов выражения энергии ускоряет процедуру её пересчета в 8-10 раз, а алгоритм в целом - в 7-8 раз.
Для оценки эффективности, предложенных в Главе 2 методов распараллеливания алгоритма моделирования диффузии, мы использовали его для поиска равновесного состояния нанесенной частицы платины размером в 4033 атома при температуре 1160 К. При моделировании использовалось от 1 до 8 ядер процессора Intel Core 2 Duo. Как видно из графика 4, использование параллельных попыток даёт лучший результат, особенно до двух потоков. Но метод пространственной декомпозиции лучше масштабируется и показывает плавный прирост производительности при увеличении числа ядер. Нужно отметить, что, несмотря на рост числа успешных актов диффузии, метод пространственной декомпозиции теряет свою эффективность из-за уменьшения размера независимых областей. Для частицы из 4033 атомов, этот метод выгодно использовать до 4-х потоков. Масштабируемость метода параллельных попыток ограничивается ростом числа одновременных успешных шагов. Судя по графику 4, при температуре 1160 К этот метод выгодно использовать до 4-6 потоков.
Подбирая параметры взаимодействия между атомами частицы и подложки, мы получили в модели форму наночастицы, соответствующую экспериментальным изображениям - сравните рисунки 5.
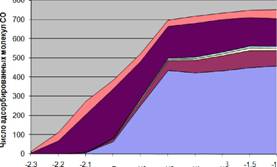
Используя разработанный алгоритм и данные об активных центрах на гранях (111) и (100), мы выполнили моделирование адсорбции на поверхности полученной частицы. Наночастица с равновесным покрытием молекулами СО приведена на рисунке 5. Для разных значений химического потенциала адсорбируемого газа (давления) было получено равновесное покрытие и распределение занятых активных центров адсорбции (рисунок 6 слева ). Полученное распределение занятых активных центров по типам atop (моноцентровая адсорбция) и
18
|
-2аа -1,9а -1,8а -1,7а -1,(
Химический потенциал ftj}, eV
- 100 bridgeаа ж 100 atop
- 111 hep ? 111 fee
ж 111 bridgeаа ? 111 atop
0.6 - |
||
s 0.4 - 2 Q. XL О 0.2 - |
||
П е1 ж |
||
2200а 2100а 2000а 1900а 1800
Частота колебаний CO на Pt{cm"1}
Рис. 6. Распределение занятых центров адсорбции по типам (слева). FTIR спектр для адсорбированного СО на нанесенных наночастицах платины (справа)
 Т~|аа Iаа ж'* I | I i I | и |" |
Т~|аа Iаа ж'* I | I i I | и |" |
0.4аа 0.Э а1 А6 каа 10 30аа 1 * 1С'7
exposure (10^ mbar Х s) т*мг
0,32аа 1 3,16
Давление, 10Е-7 мбар
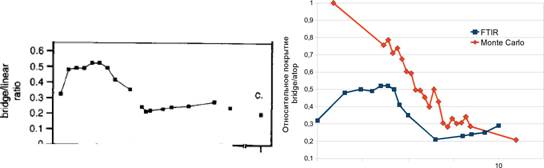
Рис. 7. Зависимость отношения числа занятых активных центров типа bridge к занятым центрам типа atop в зависимости от химического потенциала или давления. Слева - экспериментальный график из работы R. Martin et al // Surf. Sci. Ч 1995. Ч Vol. 342, no. 1-3. Ч Pp. 69-84. Справа - результаты моделирования (Monte Carlo) и приблизительное построение экспериментального графика (FTIR).
bridge (мостиковая) соответствует аналогичным данным, полученным из экспериментальных FTIR спектров: на рисунке 6 (справа) левому пику соответствуют молекулы на активных центрах типа atop, а правому - тип bridge. И FTIR спектры, и результаты моделирования говорят о том, что адсорбция типа atop более предпочтительна.
Аналогичные измерения были сделаны для грани (100) в интервале значений химического потенциала от -2.3 eV до -1.3 eV; график относительного числа занятых активных центров приведен на рисунке 7. Поскольку при моделировании мы управляли химическим потенциалом адсорбента, а не его давлением, то для того, чтобы сравнить графики, мы преобразовали химический потенциал в давление. Для этого мы использовали уравнение химического потенциала идеального газа и линейное смещение потенциала /л = 0.3// - 0.68 для того,
19
что бы компенсировать неидеальность газа и взаимодействия адсорбированных молекул. Полученный график качественно совпадает с аналогичным графиком, полученным из экспериментов. Оба графика показывают, что при низких давлениях доля мостиковой адсорбции (bridge) выше, чем при высоких, и достигает 50% от общего покрытия, но с повышением давления моноцентровая адсорбция (atop) начинает преобладать.
Таким образом, результаты моделирования и экспериментальные данные о вкладе различных типов активных центров в общее покрытие наночастиц при различном давлении, качественно совпадают, что подтверждает предсказательную силу модели.
Для достижения равновесного покрытия одной частицы понадобилось выполнить 1000 шагов Монте-Карло, что потребовало около 30 минут вычислений на процессоре Intel Core 2 Duo Е8400. Достигнутая производительность, очевидно, является приемлемой для проведения вычислительных экспериментов с использованием разработанных моделей и алгоритмов.
Основные результаты работы.
- Разработана математическая модель, описывающая адсорбцию молекул на сложных наноструктурированных поверхностях с учётом геометрии и энергетики разнообразных центров адсорбции, расположение которых определяется в ходе моделирования.
- Разработан и реализован алгоритм для решения задачи о равновесном покрытии наночастицы адсорбатом в рамках разработанной модели. Использование описания топологии решётки с помощью списка соседей и древовидной структуры данных для центров адсорбции позволило достичь приемлемой скорости расчета - несколько часов работы одного ядра современного микропроцессора для наночастицы из 4033 атомов.
- Разработаны алгоритмы, структуры данных и способы распараллеливания вычислений для решения задачи поиска равновесной формы наночастицы. Сложность расчета изменения энергии с использованием неаддитивных потенциалов снижена до О(М), что дало ускорение в 7-8 раз. Для хранения списков активных атомов и вакансий использованы структуры данных, имеющие сложность 0(1) для операций выборки, вставки и удаления. Разработанные способы распараллеливания основаны на пространственной декомпозиции и параллельной оценке нескольких актов диффузии. Вычисленные с помощью разработанного алгоритма форма и структура наночастицы соответствуют изображениям, полученным с помощью электронных микроскопов.
20
4. На основе разработанной математической модели адсорбции и с помощью разработанных компьютерных программ получено решение задачи о равновесном покрытии нанесенной платиновой наночастицы молекулами моноксида углерода с учетом реальной сложности её поверхности и разнообразия адсорбционных центров. Показано, что при низком давлении большая часть молекул СО адсорбируется на мостиках (bridge), но с увеличением давления и общего покрытия начинает преобладать моноцентровая адсорбция (atop). Эти результаты качественно совпадают с экспериментальными данными FTIR-спектроскопии.
Публикации автора по теме диссертации
В изданиях, рекомендованных ВАК:
1. Monte Carlo model of CO adsorption on supported Pt nanoparticle / A. Myshlyavtsev, P. Stishenko II Applied Surface Science.Ч 2010.Ч Vol. 256, no. 17.ЧPp. 5376 Ч 5380.
В других изданиях:
- Influence of spillover on equlibrium coverage of supported Pt nanoparticles by carbon monoxide: Monte Carlo simulation / A.V. Myshlyavtsev, P.V. Stishenko // XIX International Conference on Chemical Reactors CHEMREACTOR-19. Abstracts. - Novosibirsk: Boreskov Institute of Catalysis, 2010- Pp. 565-566.
- Различные подходы к распараллеливанию алгоритма моделирования металлических наночастиц методом Монте-Карло / Стишенко П.В. // Параллельные вычислительные технологии (ПаВТ'2010): Труды международной научной конференции (Уфа, 29 марта - 2 апреля 2010 г.). Ч Челябинск: Издательский центр ЮУрГУ, 2010. Ч С. 683.
- Monte Carlo simulation of CO adsorption on supported Pt nanoparticles / A. Myshlyavtsev, P. Stishenko // The Seventh International Symposium on Effects of Surface Heterogeneity in Adsorption and Catalysis on Solids ISSHAC-7, Proceedings, 5 July - 11 July, 2009. - Poland: Kazimierz Dolny, 2009. - P. 138-140.
- Имитационное моделирование наночастиц с использованием неаддитивных многочастичных потенциалов / Мышлявцев А.В., Стишенко П.В. // Труды 51-й научной конференции МФТИ Современные проблемы фундаментальных и прикладных наук: Часть IV. Молекулярная и биологическая физика. - М.: МФТИ, 2008. Ч С. 134-136.
21
- Monte Carlo simulation of adsorption on supported nanoparticles / A. Myshlyavtsev, P. Stishenko // XVIII International Conference on Chemical Reactors CHEMREACTOR-18. Abstracts. - Novosibirsk: Boreskov Institute of Catalysis, 2008. Ч Pp. 77-78.
- Nanoparticles simulation with non-lattice model: Monte Carlo method / A. V. Myshlyavtsev, P. V. Stishenko // Tenth International Symposium on Heterogeneous Catalysis: Quo Vadis, Catalysis? 23 - 27 August 2008, Varna, Bulgaria.
- Sofia: Institute of Catalysis Bulgarian Academy of Sciences, 2008. - P. 53.
- Использование метода Монте-Карло для моделирования частиц наномет-рового размера на примере сплава железа и платины / Мышлявцев А.В., Стишенко П.В. // Омский научный вестник.Ч 2007. Ч №(3)60. Ч С. 17-20.
- Влияние подложки на сегрегацию биметаллических частиц нанометрово-го размера: метод Монте-Карло / Стишенко П.В. // IV всероссийская научная молодежная конференция Под знаком ?. Тезисы докладов. - Омск.
Ч 2007.-С. 128-129.
Свидетельства об официальной регистрации программных систем разработанных на основе результатов диссертации
1. Monte Carlo simulation tool for metallic nanoparticles, per. номер 2009613884 (17.07.2009) / Мышлявцев A.B., Стишенко П.В. // Программы для ЭВМ. Базы данных. Топологии интегральных микросхем.Ч 2009. Ч №.4(69). ЧС. 85.
В совместных работах автор диссертации принимал участие в постановке вычислительной задачи и анализе вычислительной сложности математических моделей, выборе и разработке алгоритмов и структур данных, анализе достоверности моделей с точки зрения соответствия экспериментальным данным. В работе для Applied Surface Science автором делался обзор современных работ по моделированию адсорбции методом теории функционала плотности. В работе для 51-й научной конференции МФТИ автор делал обзор литературы на предмет описания вычислительной сложности различных эмпирических потенциалов. Автор также реализовывал все необходимые алгоритмы на языке программирования C++.
22
Авторефераты по темам >>
Разные специальности - [часть 1] [часть 2]